很多朋友都有这样的疑问,为什么别人绘制出来的热图,差异那么明显,除了首先他们本身就先做了差异分析,挑选出来了有差异的基因,然后才热图可视化外,其实还有一个步骤,就是按照基因(行)对表达矩阵进行zscore转换。
关于差异分析的细节,大家可以自行细读表达芯片的公共数据库挖掘系列推文 ;
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
我这里就不赘述啦。
我们直接来看作图细节:
首先看原始表达矩阵热图
代码如下:
# 2.热图
load(file='heatmap_input.Rdata')
## 2.1 数据预处理
t <- log2(cgexp+1)
t <- na.omit(t) #取出为空(表达矩阵中没有)的基因
dim(t)
## 准备画图
library(pheatmap) #加载包
identical(colnames(t),colnames(mRNA_exp_small)) #确定一下顺序没变,方便后面添加分组信息
### 构建注释矩阵
col <- data.frame(Type=group_list) #显示肿瘤类型
col$Type <- factor(col$Type,levels = c('normal','tumor'))
rownames(col) <- colnames(t)
### 开始画图
如下:
![原始表达矩阵热图]http://www.bio-info-trainee.com/wp-content/uploads/2020/05/image-20200517204957059.png)
{kind=link}
可以看到,两个分组差异是有的,但是肉眼其实看不清楚基因层面哪些高表达哪些低表达。因为不同基因的表达矩阵本身差异很大,但其实我们仅仅是关心同一个基因在不同分组样本的表达,我们并不会关系不同基因的表达量问题,所以需要按照基因(行)对表达矩阵进行zscore转换。
第一次zscore
代码如下:
## 2.2 进行scale,但是不设定最大最小值
t <- t(scale(t(t)))
p1 <- pheatmap(t,
annotation_col = col,
annotation_legend = T,
border_color = 'black',#设定每个格子边框的颜色,border=F则无边框
cluster_rows = T, #对行聚类
cluster_cols = T, #队列聚类
show_colnames = F, #是否显示列名
show_rownames = F #是否显示行名
)
出图如下:
![第一次zscore]http://www.bio-info-trainee.com/wp-content/uploads/2020/05/image-20200517205144147.png)
{kind=link}
可以看到,上下调的基因清晰可见,但是这个时候很多人不理解热图上面的层次聚类,会错误的以为tumor被normal隔离成为了两个分组。
这个时候,如果你使用我的热图代码,通常是会在zcore的时候,设置一个上限值,比如2或者1.6,代码如下:
然后限定zscore的范围
代码如下:
## 2.3 进行scale后设定最大最小值的情况
table(abs(t)>2)
t[t>=2]=2
t[t<=-2]=-2
table(is.na(t))
p2 <- pheatmap(t,
annotation_col = col,
annotation_legend = T,
border_color = 'black',#设定每个格子边框的颜色,border=F则无边框
cluster_rows = T, #对行聚类
cluster_cols = T, #队列聚类
show_colnames = F, #是否显示列名
show_rownames = F #是否显示行名
)
出图如下:
![限定zscore的范围]http://www.bio-info-trainee.com/wp-content/uploads/2020/05/image-20200517205251593.png)
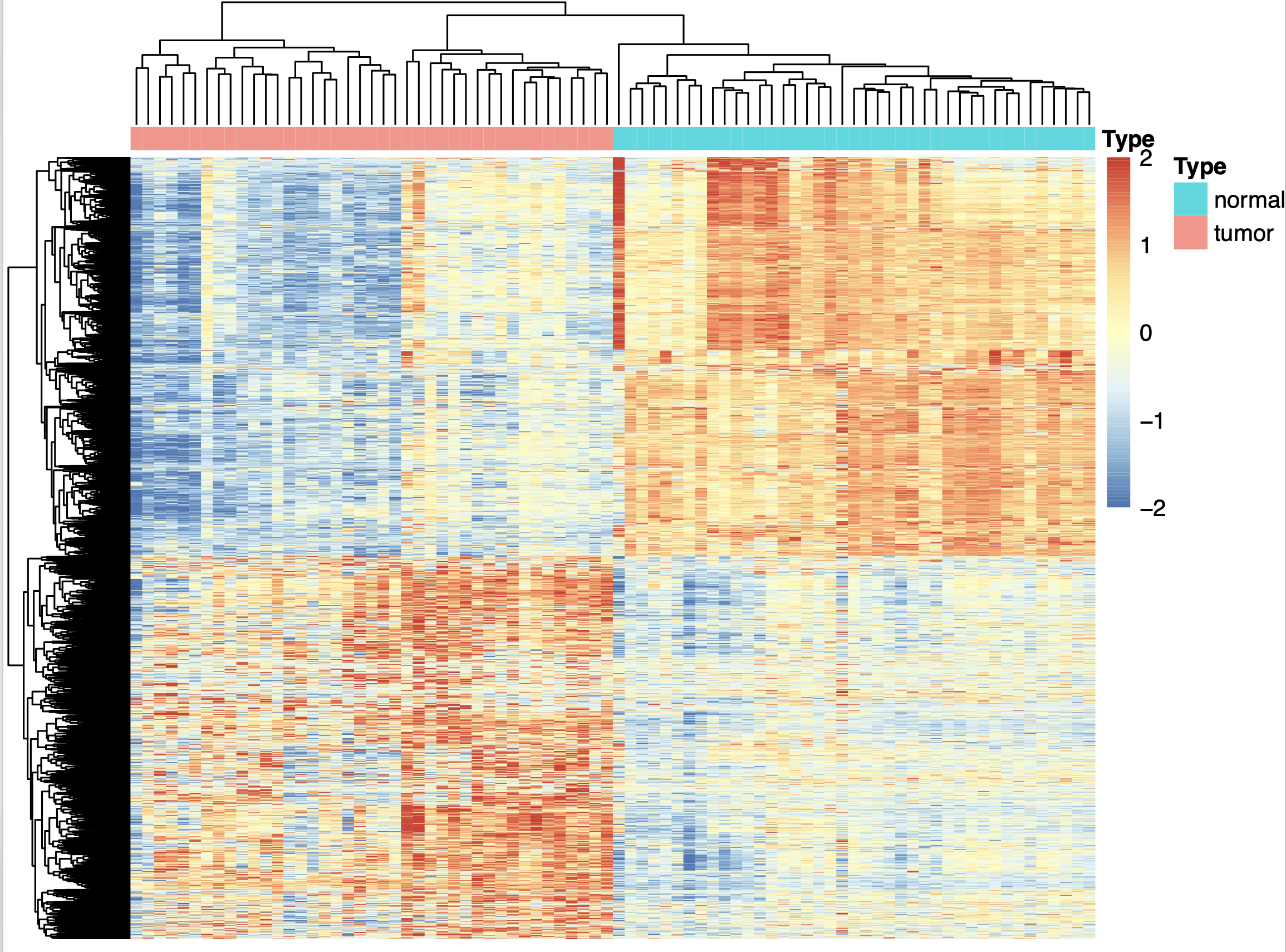{kind=link}
很有意思,这个时候上下调基因仍然是清晰可见,很容易看出来高低表达量分组,而且不会出现上面热图的tumor被normal隔离成为了两个分组的假象!
那么问题来了,这样是操纵数据吗
是不是很有意思,有时候你很难给合作者解释清楚。