shell
echo "START"
大家好,我是熊猫。
事情是这样的,前些天我在朋友圈发了一张图片:
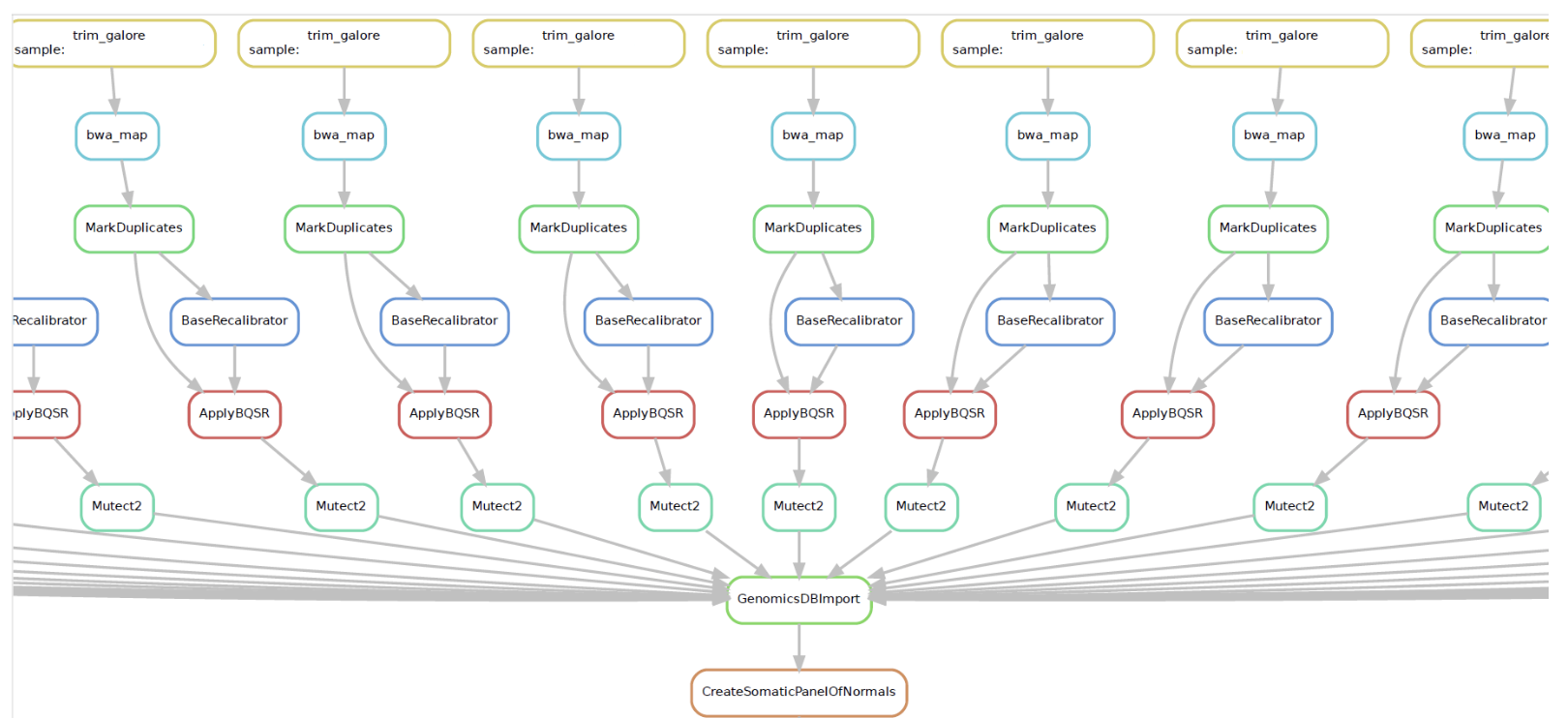
这是使用gatk4生成正常样本的germline突变数据库的流程图,整个流程是用Snakemake写的,这个图片也是Snakemake生成的。然后就被jimmy大佬点名了,受宠若惊,所以就有了本文。我是2016年从转录组学习小分队开始正式接触生信技能树,并走上了生信工程师的道路,我被jimmy大佬无私奉献的精神所折服,借此机会表示对jimmy大佬和生信技能树由衷的感谢!如果你也想从转录组开启你的生物信息学学习之旅,不妨考虑一下生信技能树的爆款入门:
本文主题为使用Snakemake搭建生信分析流程,下面开始正文。
准备工作
正式开始前,你需要完成以下工作:
1、在linux环境下安装好conda,并使用conda安装好gatk4(4.1.6.0)、Snakemake(5.13.0)、trim-galore(0.6.5)、 bwa(0.7.17)。关于生物信息学环境搭建的讨论,大家可以看生信菜鸟团专题:
关于conda本身学习可以看 :conda管理生信软件一文就够 但是要理解软件安装原理,就需要脱离conda练习安装软件,比如生物信息学常见1000个软件的安装代码!
2、了解gatk4的数据预处理流程(Data pre-processing for variant discovery)和生成正常样本的germline突变数据库的流程(A step-by-step guide to the new Mutect2 Panel of Normals Workflow)。
Snakemake的使用
Snakemake是基于Python写的流程管理软件,我理解为一个框架。Snakemake的基本组成单位是rule,表示定义了一条规则。每一个rule包含三个基本元素,分别是input、output、shell或run或script,分别表示“输入文件”、“输出文件”和“运行命令”。Snakemake会自动判断一条rule的input是来自哪条rule的output,从而将一条条rule串成一个完整的流程。这是Snakemake的一个优点,另外Snakemake支持“断点续行”,假如你的任务运行到一半因为某种原因中断了,你可以重新运行一下命令,Snakemake会机智的从中断的地方继续运行,已经成功运行的任务不会重复运行;Snakemake支持并行处理任务,可以设定运行核心数或并行任务数,也可以将任务投递到集群运行。
我用到的文件和对应的路径(需要自己准备到服务器,测试数据和软件依赖的数据库文件)
├── sample1
│ ├── sample1.L1-B1.R1.fastq.gz
│ └── sample1.L1-B1.R2.fastq.gz
├── sample2
│ ├── sample2.L1-B1.R1.fastq.gz
│ └── sample2.L1-B1.R2.fastq.gz
├── sample3
│ ├── sample3.L1-B1.R1.fastq.gz
│ └── sample3.L1-B1.R2.fastq.gz
├── database
│ ├── hg19.fa
│ ├── hapmap_3.3.hg19.sites.vcf
│ ├── 1000G_omni2.5.hg19.sites.vcf
│ ├── dbsnp_138.hg19.sites.vcf
│ ├── 1000G_phase1.indels.hg19.sites.vcf
│ ├── Mills_and_1000G_gold_standard.indels.hg19.sites.vcf
│ └── af-only-gnomad.raw.sites.hg19.vcf.gz
├── bed
│ └── Covered.bed
├── config.yaml
└── Snakefile
新建一个配置文件config.yaml
内容和格式为:
samples:
sample1:
sample2:
sample3:
新建一个流程文件Snakefile
首先定义配置文件config.yaml
configfile: "config.yaml"
Snakemake读取配置文件后会将数据保存为字典,这是一个简单的示范,配置文件也可以写的复杂,比如定义每个样本所用的bed文件或不同的分析参数。获取样本列表的方式为:sample=config["samples"]。
然后是定义最终需要的结果文件:
rule all:
input:
"gatk4_mutect2_pon.vcf.gz"
all是每个Snakefile文件中必有的一个rule,比较特殊,只需要一个input,用来定义流程最终输出的结果。注意:如果你的流程有不同的分支,最终会生成多个需要的结果,那么这些结果都需要在这里定义。
接下来是构建分析流程
第一步是去接头(trim_galore):
rule trim_galore:
input:
"{sample}/{sample}.L1-B1.R1.fastq.gz",
"{sample}/{sample}.L1-B1.R2.fastq.gz"
output:
"{sample}/clean_fq/{sample}.L1-B1.R1_val_1.fq.gz",
"{sample}/clean_fq/{sample}.L1-B1.R2_val_2.fq.gz"
shell:
"trim_galore --paired --retain_unpaired -q 20 --phred33 --length 30 --stringency 3 --gzip --cores 4 -o {wildcards.sample}/clean_fq {input}"
input样本目录下的两个fastq文件,output为样本目录下clean_fq文件夹下的两个去过接头的fastq文件,shell里就是我们平常写的shell命令,只不过可以把输入文件和输出文件用input和output替代。这里需要注意:1、Snakemake会自动创建不存在的目录;2、如果shell命令没有定义输出文件,也可以不写output;3、这一步使用了{sample}这个参数,但实际上{sample}还没有定义,单独运行这一步会报错,{sample}的定义在后面的rule。另外,如果在shell中要使用这个参数,还需要加上wildcards,即{wildcards.sample}。
第二步,bwa比对(bwa_map):
rule bwa_map:
input:
"database/hg19.fa",
"{sample}/clean_fq/{sample}.L1-B1.R1_val_1.fq.gz",
"{sample}/clean_fq/{sample}.L1-B1.R2_val_2.fq.gz"
output:
"{sample}/{sample}.mem.bam"
params:
rg=r"@RG\tID:{sample}.L1-B1\tSM:{sample}\tLB:L1-B1\tPL:ILLUMINA"
shell:
"bwa mem \
-M -Y \
-R '{params.rg}' \
-t 16 \
{input} | \
samtools view -1 - > {output}"
这一步用到了params,在这里定义命令中用到的参数,也可以直接从配置文件中读取。如果你的shell命令中有双引号,需要使用\进行转义或者使用单引号。
第三步,标记重复序列(MarkDuplicates):
rule MarkDuplicates:
input:
"{sample}/{sample}.mem.bam"
output:
bam = "{sample}/{sample}.markdup.bam",
txt = "{sample}/{sample}.markdup_metrics.txt"
params:
aso = r"queryname",
so = r"coordinate"
shell:
"gatk MarkDuplicates \
--INPUT {input} \
--OUTPUT /dev/stdout \
--METRICS_FILE {output.txt} \
--VALIDATION_STRINGENCY SILENT \
--OPTICAL_DUPLICATE_PIXEL_DISTANCE 2500 \
--ASSUME_SORT_ORDER '{params.aso}' \
--CREATE_MD5_FILE false \
--REMOVE_DUPLICATES false | \
gatk SortSam \
--INPUT /dev/stdin \
--OUTPUT {output.bam} \
--SORT_ORDER '{params.so}' \
--CREATE_INDEX false \
--CREATE_MD5_FILE false"
第四步,碱基质量分数重校准(BaseRecalibrator):
rule BaseRecalibrator:
input:
"{sample}/{sample}.markdup.bam"
output:
"{sample}/{sample}.bqsr_data.table"
shell:
"gatk BaseRecalibrator \
-R database/hg19.fa \
-I {input} \
--use-original-qualities \
-O {output} \
--known-sites database/hapmap_3.3.hg19.sites.vcf \
--known-sites database/1000G_omni2.5.hg19.sites.vcf \
--known-sites database/dbsnp_138.hg19.sites.vcf \
--known-sites database/1000G_phase1.indels.hg19.sites.vcf \
--known-sites database/Mills_and_1000G_gold_standard.indels.hg19.sites.vcf"
第五步,应用碱基质量分数重校准(ApplyBQSR):
rule ApplyBQSR:
input:
bam = "{sample}/{sample}.markdup.bam",
table = "{sample}/{sample}.bqsr_data.table"
output:
"{sample}/{sample}.bqsr.bam"
shell:
"gatk ApplyBQSR \
-R database/hg19.fa \
-I {input.bam} \
-O {output} \
-bqsr {input.table} \
--static-quantized-quals 10 --static-quantized-quals 20 --static-quantized-quals 30 \
--add-output-sam-program-record \
--create-output-bam-index true \
--create-output-bam-md5 true \
--use-original-qualities"
第六步,Mutect2找变异(Mutect2):
rule Mutect2:
input:
"{sample}/{sample}.bqsr.bam"
output:
"{sample}/{sample}.vcf.gz"
shell:
"gatk Mutect2 \
-R database/hg19.fa \
-I {input} --max-mnp-distance 0 \
-O {output}"
第七步,基因组数据库导入(GenomicsDBImport):
rule GenomicsDBImport:
input:
expand("{sample}/{sample}.vcf.gz", sample=config["samples"])
output:
directory("pon_db")
params:
" -V ".join(expand("{sample}/{sample}.vcf.gz", sample=config["samples"]))
shell:
"gatk GenomicsDBImport \
-R database/hg19.fa \
--genomicsdb-workspace-path {output} \
-V {params} \
-L bed/Covered.bed"
这一步需要用到所有样本的vcf文件,使用python的expand命令将每个样本的vcf文件依次添加到一个列表中。在这里定义了参数sample,Snakemake从rule all回溯到这里的时候就知道了sample代表的具体样本名。如果output定义的是一个目录,需要加上directory;相反如果input定义的是一个目录,就不需要加directory。
第八步,创建正常样本的数据库(CreateSomaticPanelOfNormals):
rule CreateSomaticPanelOfNormals:
input:
"pon_db"
output:
"gatk4_mutect2_pon.vcf.gz"
shell:
"gatk CreateSomaticPanelOfNormals -R database/hg19.fa \
--germline-resource database/af-only-gnomad.raw.sites.hg19.vcf.gz \
-V gendb://{input} \
-O {output}"
到这里流程全部搭建完成,保存Snakefile文件。运行命令snakemake --dag | dot -Tpdf > dag.pdf就可以生成本文开头的流程图。运行命令snakemake -np可以预览所有的shell命令。通过添加--cores/--jobs/-j N参数可以指定并行数,如果不指定N,则使用当前最大可用的核心数。一切准备妥当,运行命令snakemake --cores 16,程序就跑起来了。
扩展
rule中还可以添加其他的参数,比如说threads、log,如果输出文件重要,可以添加protected参数设置为保护文件,相反,如果跑完程序就可以删除的文件,可以添加temp参数设置为临时文件。
rule NAME:
input:
"path/to/inputfile",
"path/to/other/inputfile"
output:
protected("path/to/outputfile"),
temp("path/to/another/outputfile")
log:
"logs/abc.log"
threads:
8
shell:
"somecommand --threads {threads} {input} {output}"
更多用法请前往Snakemake的官方文档。如有问题,欢迎交流和指正。
echo "DONE"
文末友情宣传
强烈建议你推荐我们生信技能树给身边的博士后以及年轻生物学PI,帮助他们多一点数据认知,让科研更上一个台阶:
- 生信爆款入门-全球听(买一得五)(第4期),你的生物信息学入门课
- 数据挖掘第2期(两天变三周,实力加量),医学生/临床医师首选技能提高课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路,还等什么,看啊!!!