号外:绝大部分生信技能树粉丝都没有机会加我微信,已经多次满了5000好友,所以我开通了一个微信好友,前100名添加我,仅需150元即可,3折优惠期机会不容错过哈。我的微信小号二维码在:0元,10小时教学视频直播《跟着百度李彦宏学习肿瘤基因组测序数据分析》
我已经在生信技能树多次介绍过生存分析,目录如下
- 集思广益-生存分析可以随心所欲根据表达量分组吗
- 生存分析时间点问题
- 寻找生存分析的最佳基因表达分组阈值
- apply家族函数和for循环还是有区别的(批量生存分析出图bug)
- TCGA数据库生存分析的网页工具哪家强
现在有了《专辑》这个功能,其实更方便查看我们的历史教程啦。因为我五年前做生存分析研发这个代码的时候,就是根据基因表达量,把病人分成了高低表达两个组,不管是使用cox还是km,都是这样做的。但是最近有学生反映,使用cox还是km拿到的基因的生存效果是一致的, 就是风险因子和保护因子的分类问题。主要是靠HR值来判断咯。
关于HR值
In summary,
- HR = 1: No effect
- HR < 1: Reduction in the hazard
- HR > 1: Increase in Hazard
Note that in cancer studies:
- A covariate with hazard ratio > 1 (i.e.: b > 0) is called bad prognostic factor
- A covariate with hazard ratio < 1 (i.e.: b < 0) is called good prognostic factor
其中KM方法代码是
大家需要自行进行数据清洗,拿到表达矩阵和临床信息哦,其中临床信息还需要有time, event这两列哦。然后对表达矩阵里面的全部基因,根据其表达量高低把病人区分成为两组后,批量走生存分析拿到结果。
## 批量生存分析 使用 logrank test 方法
mySurv=with(phe,Surv(time, event))
log_rank_p <- apply(exprSet,1,function(gene){
# gene=exprSet[1,]
phe$group=ifelse(gene>median(gene),'high','low')
data.survdiff=survdiff(mySurv~group,data=phe)
p.val = 1 - pchisq(data.survdiff$chisq, length(data.survdiff$n) - 1)
return(p.val)
})
log_rank_p=sort(log_rank_p)
head(log_rank_p)
boxplot(log_rank_p)
table(log_rank_p<0.05)
p <- log_rank_p[log_rank_p<0.05]
其中cox方法代码是:
代码略微复杂一点,但都是根据基因的表达量中位值,把病人区分成为高低表达量的两个组,然后进行生存分析检验。
## 批量生存分析 使用cox 方法
mySurv=with(phe,Surv(time, event))
identical(substring(colnames(exprSet),1,12), phe$ID)
cox_results <-apply(exprSet , 1 , function(gene){
# gene= exprSet[1,]
group=ifelse(gene>median(gene),'high','low')
survival_dat <- data.frame(group=group,
stringsAsFactors = F)
m=coxph(mySurv ~ group, data = survival_dat)
beta <- coef(m)
se <- sqrt(diag(vcov(m)))
HR <- exp(beta)
HRse <- HR * se
#summary(m)
tmp <- round(cbind(coef = beta, se = se, z = beta/se, p = 1 - pchisq((beta/se)^2, 1),
HR = HR, HRse = HRse,
HRz = (HR - 1) / HRse, HRp = 1 - pchisq(((HR - 1)/HRse)^2, 1),
HRCILL = exp(beta - qnorm(.975, 0, 1) * se),
HRCIUL = exp(beta + qnorm(.975, 0, 1) * se)), 3)
return(tmp['grouplow',])
})
cox_results=t(cox_results)
但是学生使用了cox的连续变量
就是不需要根据基因表达量把病人分组, 直接就把基因表达量相关连续变量代入cox代码里面,如下:
mySurv=with(phe,Surv(time, event))
identical(substring(colnames(exprSet),1,12), phe$ID)
cox_2 <-apply(exprSet , 1 , function(gene){
# gene= as.numeric(exprSet[1,])
x=coxph(mySurv ~ log(gene+1) )
x <- summary(x)
p.value<-signif(x$wald["pvalue"], digits=2)
wald.test<-signif(x$wald["test"], digits=2)
beta<-signif(x$coef[1], digits=2);#coeficient beta
HR <-signif(x$coef[2], digits=2);#exp(beta)
HR.confint.lower <- signif(x$conf.int[,"lower .95"], 2)
HR.confint.upper <- signif(x$conf.int[,"upper .95"],2)
HRm <- paste0(HR, " (",
HR.confint.lower, "-", HR.confint.upper, ")")
res<-c(beta, HR,HR.confint.lower,HR.confint.upper, wald.test, p.value)
names(res)<-c("beta",'HR','HR.confint.lower','HR.confint.upper', "wald.test",
"p.value")
res
})
cox_2=t(cox_2)
比较两次cox的结果
超级简单的绘图如下:
colnames(cox_results)
colnames(cox_2)
plot(cox_results[,5],cox_2[,2])
cor(cox_results[,5],cox_2[,2])
如图:
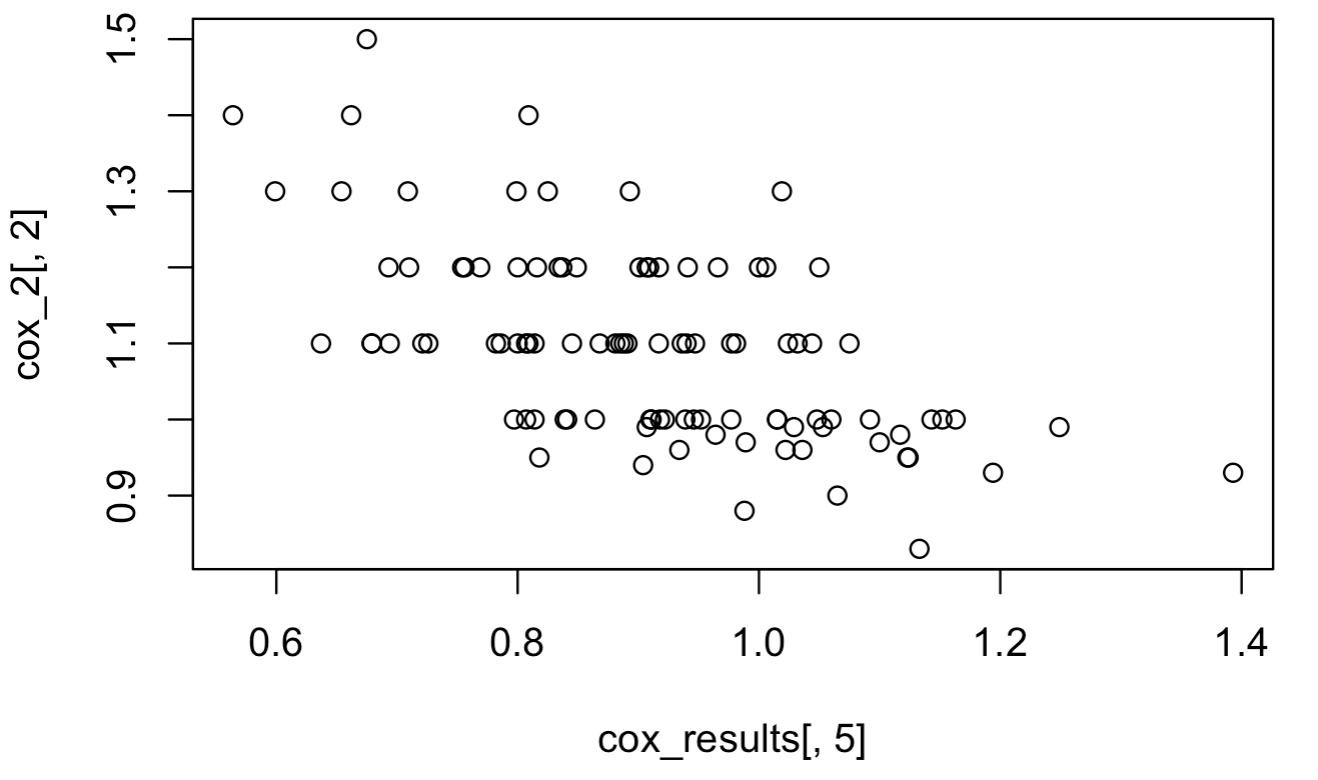
很明显,两次cox结果是负相关。
实际上,这就是一个文字游戏。
我们说A基因是风险因子,那么意思是,具有A基因高表达这个现象的病人理论上预后更差。反应在生存分析曲线图上面,就是如下:
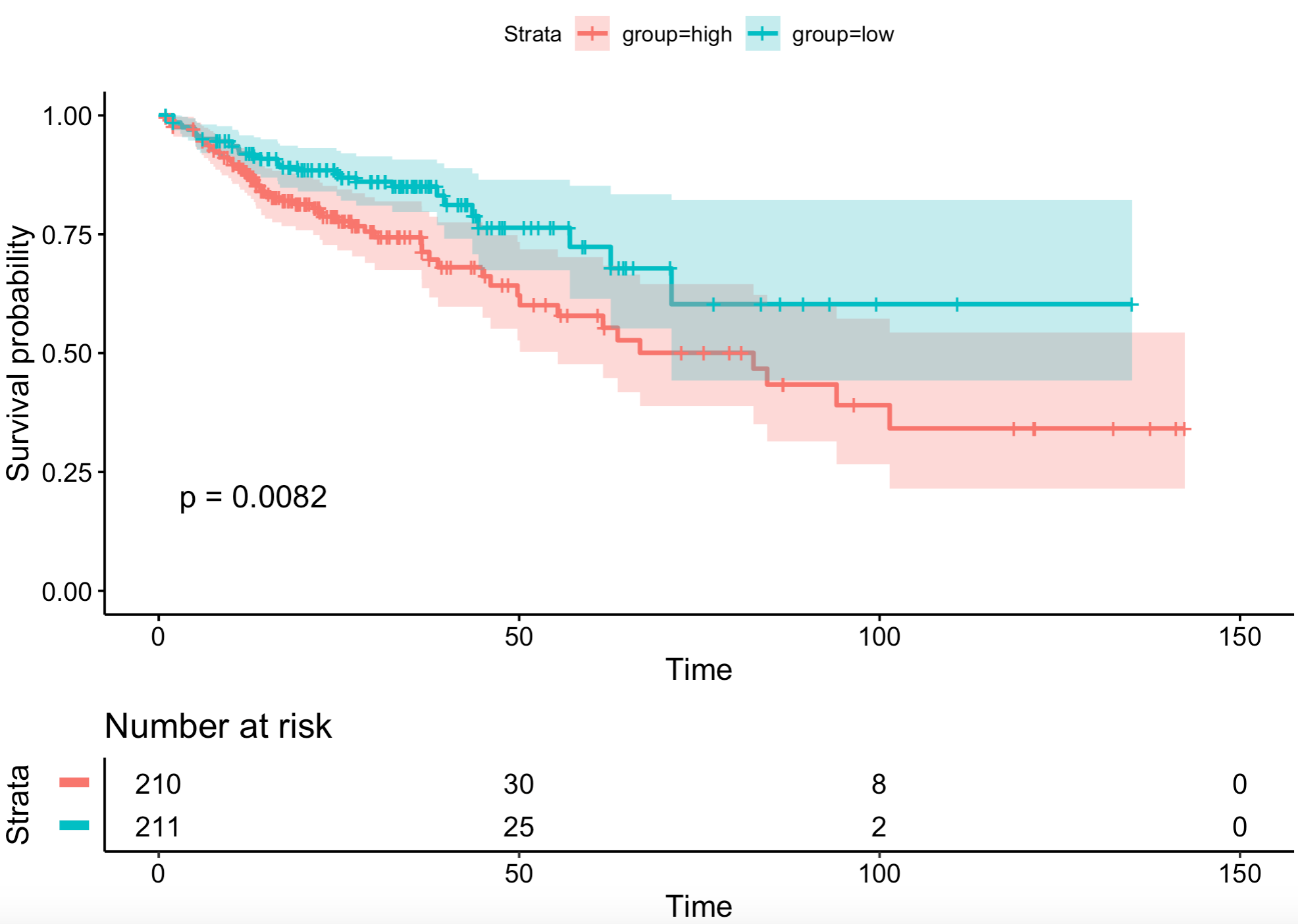
如果我们经过km的logRANK或者cox的分类或者连续变量的生存分析检验,计算出来该基因的HR是小于1的,说明它是降低风险,理论上是保护因子,在曲线上面会反映出来的是靠下。
但是我们前面的cox分类生存分析后,拿到的是return(tmp[‘grouplow’,])的各个计算结果,意思是该基因的低表达才是保护因子, 高表达量的它是风险因素。
所以cox的连续变量计算出来的HR值,就全部反过来了。