有粉丝邮件求助,给了我两个vcf文件,旧的vcf文件走的是标准的bwa+gatk流程,参考基因组是hg19,新的文件参考基因组是hg38,也是gatk标准流程。想要比较它们,首先得保证两个vcf文件的参考基因组一致,因为版本不一致,所以需要使用CrossMap等软件进行参考基因组版本转换,然后使用 SnpSift 软件的 Concordance 命令比较它们。
如果简单看文件数量,当然是不行的。
137352 new.filter.sort.vcf
46976 old.filter.sort.vcf
首先看看两个vcf文件的染色体分布情况
cat old.filter.sort.vcf |grep -v '^#' |cut -f 1 |sort |uniq -c > old.chr
cat new.filter.sort.vcf |grep -v '^#' |cut -f 1 |sort |uniq -c > new.chr
paste new.chr old.chr
结果如下:
14341 chr1 4912 chr1
6562 chr10 2108 chr10
7427 chr11 2920 chr11
7436 chr12 2519 chr12
3200 chr13 856 chr13
4051 chr14 1408 chr14
4568 chr15 1520 chr15
5934 chr16 2223 chr16
7100 chr17 2702 chr17
2596 chr18 778 chr18
8020 chr19 3399 chr19
10160 chr2 3146 chr2
3349 chr20 1135 chr20
1908 chr21 666 chr21
3262 chr22 1095 chr22
7676 chr3 2708 chr3
6437 chr4 1990 chr4
6134 chr5 1975 chr5
6265 chr6 2718 chr6
7095 chr7 2115 chr7
4813 chr8 1595 chr8
6162 chr9 1915 chr9
31 chrM
2081 chrX 519 chrX
157 chrY 7 chrY
可以看到,新的vcf文件的突变位点数量远大于旧的vcf文件。仔细查看新vcf文件,发现是没有做基本过滤,比如测序深度大于20等等指标。所以我就顺便把它过滤一波,代码如下:
input=new.filter.sort.vcf
cat $input | java -jar ~/biosoft/snpEff/SnpSift.jar filter "( DP > 20 & FILTER = 'PASS' )" | \
perl -alne '{print unless $F[0] =~ /_/}' | \
awk '$1 ~ /^#/ {print $0;next} {print $0 | "sort -k1,1 -k2,2n"}' | \
grep -v '1/2' > filtered.chr
# 然后再统计这个vcf文件的染色体发布情况
cat filtered.vcf |grep -v '^#' |cut -f 1 |sort |uniq -c > filtered.chr
paste new.chr old.chr filtered.chr
如下所示:
14341 chr1 4912 chr1 5731 chr1
6562 chr10 2108 chr10 2617 chr10
7427 chr11 2920 chr11 3419 chr11
7436 chr12 2519 chr12 2971 chr12
3200 chr13 856 chr13 1070 chr13
4051 chr14 1408 chr14 1636 chr14
4568 chr15 1520 chr15 1780 chr15
5934 chr16 2223 chr16 2555 chr16
7100 chr17 2702 chr17 3216 chr17
2596 chr18 778 chr18 957 chr18
8020 chr19 3399 chr19 3960 chr19
10160 chr2 3146 chr2 3822 chr2
3349 chr20 1135 chr20 1448 chr20
1908 chr21 666 chr21 722 chr21
3262 chr22 1095 chr22 1354 chr22
7676 chr3 2708 chr3 3060 chr3
6437 chr4 1990 chr4 2405 chr4
6134 chr5 1975 chr5 2334 chr5
6265 chr6 2718 chr6 2376 chr6
7095 chr7 2115 chr7 2615 chr7
4813 chr8 1595 chr8 1897 chr8
6162 chr9 1915 chr9 2497 chr9
2081 chrX 519 chrX 552 chrX
157 chrY 7 chrY 6 chrY
可以看到,经过质控后的两个vcf文件,至少是从染色体上变异位点记录的数量上来说,非常类似的。后面我们还需要进行更细致的探索。
再看基础的vcf统计命令
使用snpEff可以对vcf进行基础统计,它也会对染色体分布情况进行统计,更重要的有丰富的可视化图表
java -jar ~/biosoft/snpEff/snpEff.jar GRCh38.86 \
old.filter.sort.vcf > old.filter.sort.eff.vcf
java -jar ~/biosoft/snpEff/snpEff.jar GRCh38.86 \
new.filter.sort.vcf > new.filter.sort.eff.vcf
结果如下:
可以看到突变位点区域分类情况:
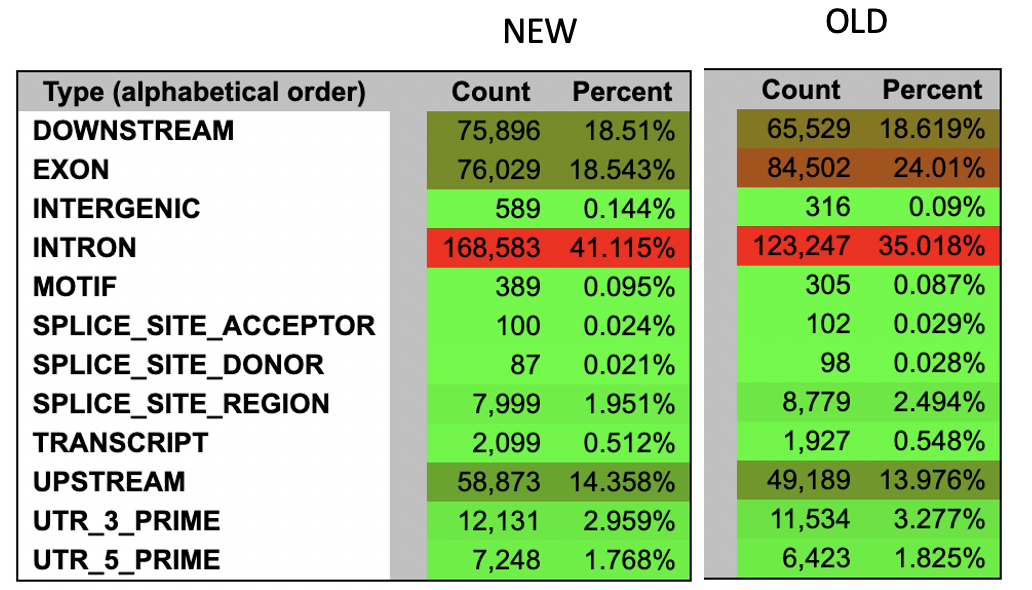
可以看到,两个vcf文件的变异位点在intron和exon区域的比例差异是最大的,其实是因为它们两个区域本来就长度很大。
另外一个统计指标
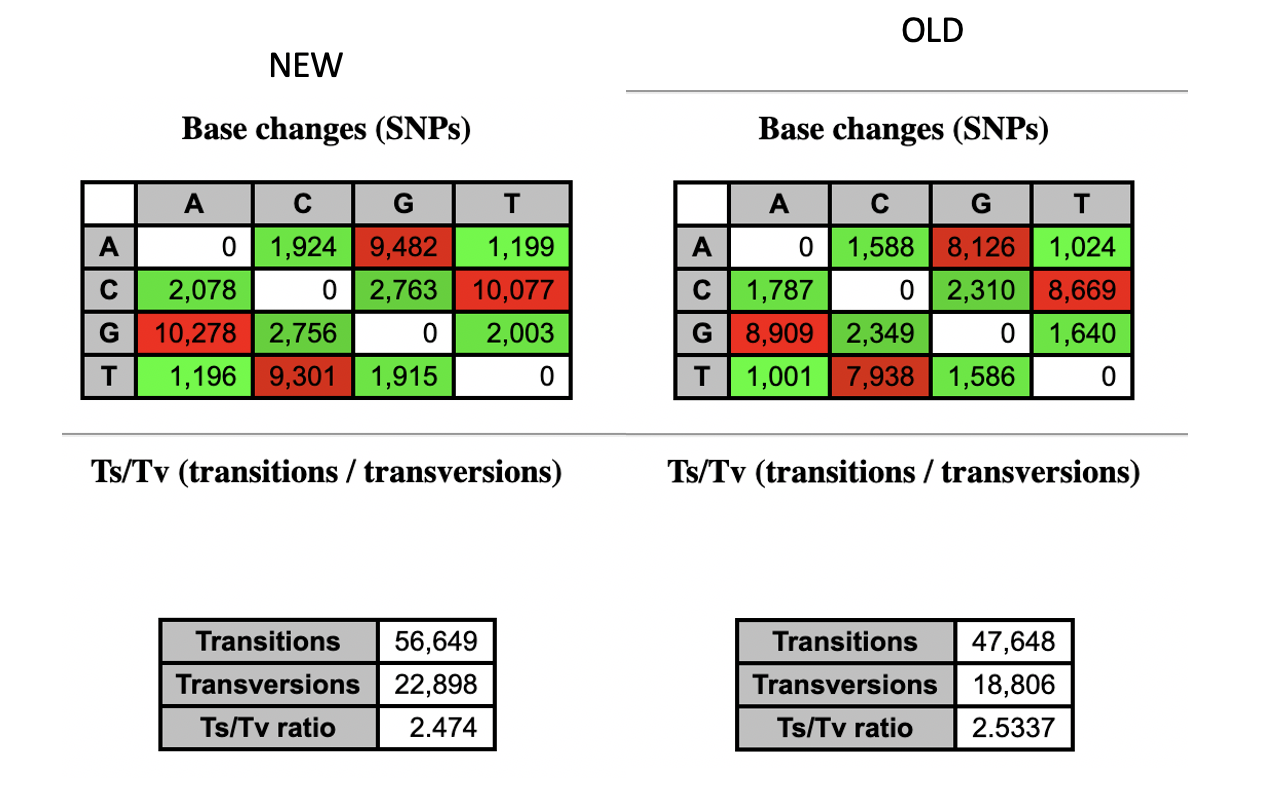
最后看专业的软件进行两个vcf文件比较
这里使用 SnpSift 软件的 Concordance 命令,代码如下:
java -Xmx1g -jar ~/biosoft/snpEff/SnpSift.jar concordance -v \
new.filter.sort.vcf old.filter.sort.vcf 1>comp.results.txt 2>comp.log.txt
得到的结果文件不是很好理解:
2.2K Jul 11 11:57 comp.log.txt
4.2M Jul 11 11:57 comp.results.txt
590 Jul 11 11:57 concordance_new_old.by_sample.txt
89 Jul 11 11:57 concordance_new_old.summary.txt
其中 concordance_new_old.by_sample.txt 文件需要倒置查看:
5 MISSING_ENTRY_new/MISSING_ENTRY_old 0
6 MISSING_ENTRY_new/MISSING_GT_old 0
7 MISSING_ENTRY_new/REF 0
8 MISSING_ENTRY_new/ALT_1 4842
9 MISSING_ENTRY_new/ALT_2 1971
10 MISSING_GT_new/MISSING_ENTRY_old 0
11 MISSING_GT_new/MISSING_GT_old 0
12 MISSING_GT_new/REF 0
13 MISSING_GT_new/ALT_1 0
14 MISSING_GT_new/ALT_2 0
15 REF/MISSING_ENTRY_old 0
16 REF/MISSING_GT_old 0
17 REF/REF 0
18 REF/ALT_1 0
19 REF/ALT_2 0
20 ALT_1/MISSING_ENTRY_old 7855
21 ALT_1/MISSING_GT_old 0
22 ALT_1/REF 0
23 ALT_1/ALT_1 22538
24 ALT_1/ALT_2 4
25 ALT_2/MISSING_ENTRY_old 7001
26 ALT_2/MISSING_GT_old 0
27 ALT_2/REF 0
28 ALT_2/ALT_1 20
29 ALT_2/ALT_2 17552
30 ERROR 0
可以看到 ALT_1/ALT_1有22538个,ALT_2/ALT_2有17552个,说明新旧vcf文件的一致性还挺好的!
然后ALT_1/MISSING_ENTRY_old是7855个,ALT_2/MISSING_ENTRY_old是7001个,说明新的vcf文件,多找出来了近1.5万个突变位点。
而MISSING_ENTRY_new/ALT_1的4842个,MISSING_ENTRY_new/ALT_2是1971个,说明旧的vcf文件,多找出来了不到7千个突变位点。
有意思的是ALT_1/ALT_1 22538
两个流程不可能完全一致,近4万个位点在两个vcf文件里面都有,超过80%的一致性了,挺好的。
如果要具体看哪些位置是一致的,哪些是冲突的,就需要去重点理解 comp.results.txt 文件。
其实可以分染色体查看
前面的 SnpSift 软件的 Concordance 命令,最后得到的是vcf文件全部染色体的,一致性的韦恩图罢了。
但是可以继续细致的探索 comp.results.txt 文件,拆分染色体后,继续统计上面提到的6种情况发生的频次。那就出一个学徒作业吧,比较两个vcf文件,然后区分染色体绘制韦恩图。
这两个vcf文件可以是不同人的,也可以是同一个人的不同批次测序或者不同数据分析流程拿到的vcf文件。
也有很多其它轮子
比如 vcf-compare 工具,bedtools等等
实际上考验的就是Linux知识
再怎么强调生物信息学数据分析学习过程的计算机基础知识的打磨都不为过,我把它粗略的分成基于R语言的统计可视化,以及基于Linux的NGS数据处理:
Linux的6个阶段需要跨越过去 ,一般来说,每个阶段都需要至少一天以上的学习:
- 第1阶段:把linux系统玩得跟Windows或者MacOS那样的桌面操作系统一样顺畅,主要目的就是去可视化,熟悉黑白命令行界面,可以仅仅以键盘交互模式完成常规文件夹及文件管理工作。
- 第2阶段:做到文本文件的表格化处理,类似于以键盘交互模式完成Excel表格的排序、计数、筛选、去冗余、查找、切割、替换、合并、补齐,熟练掌握awk、sed、grep这文本处理的三驾马车。
- 第3阶段:元字符,通配符及shell中的各种扩展,从此linux操作不再神秘!
- 第4阶段:高级目录管理:软硬链接,绝对路径和相对路径,环境变量。
- 第5阶段:任务提交及批处理,脚本编写解放你的双手。
- 第6阶段:软件安装及conda管理,让linux系统实用性放飞自我。
很多粉丝发邮件询问我具体的软件,命令报错该如何解决,这些问题通常是输入文件,命令参数问题,还有java环境问题,太细致了,我没有空去帮大家debug哦。
如果是是Linux基础知识掌握的不牢固,其实拼命学习即可: