我看到今年(2020)3月发表在:BMC Bioinformatics 的文章:《RASflow: an RNA-Seq analysis workflow with Snakemake》,就是很简单的一个转录组流程分享,只不过是他愿意写出来英文SCI而且愿意花时间投稿。如果大家学过了我免费共享在B站的不同的数据分析视频课程,见:
- 免费视频课程《RNA-seq数据分析》
- 免费视频课程《WES数据分析》
- 免费视频课程《ChIP-seq数据分析》
- 免费视频课程《ATAC-seq数据分析》
其实如果你也是有时间,愿意耗费时间把这些流程整理成文章,就可以投稿发表哦!
我觉得这个才是生信人该做的事情,而且一个人就可以独立完成:

步骤包括: - quality control (QC) and trimming,
- mapping of reads to a reference genome (or transcriptome),
- quantification on gene (or transcript) level,
- statistical analysis of expression statistics to report genes (or transcripts) being
- differentially expressed between two predefined sets of samples, along with associated P-values or False Discovery Rate (FDR) values.
代码共享在 GitHub: https://github.com/zhxiaokang/RASflow,如果你看我三年前的[免费视频课程《RNA-seq数据分析》](https://mp.weixin.qq.com/s/LoVAPDIHI4xruvw8zDeeCw),就会发现,真的是大同小异。
作者需要照例把自己的流程跟已经发表的其它转录组数据分析流程进行了比较:
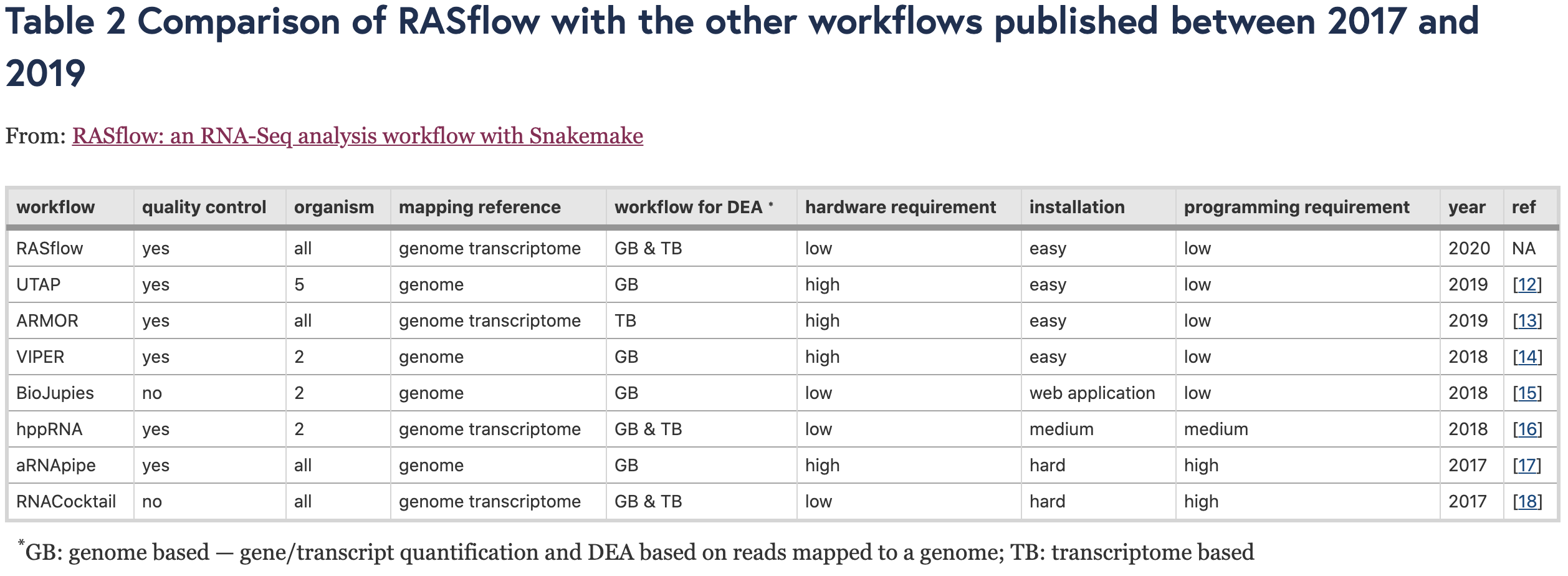
也就是说,仅仅是转录组这个领域,发表的流程相关文章就十几篇了!
不过,有意思的是流程发的再多,也是用的人也很少。但是可以非常好的锻炼到自己的能力,而且在2020年五月发表在peerJ期刊,作者有三个人,分别是Allen Hubbard,唐纳德丹佛植物科学中心(圣路易斯是美国密苏里州东部大城市),Matthew Bomhoff(美国亚利桑那大学植物与土壤科学系)和 Carl J. Schmidt(特拉华大学动物与食品科学系,纽瓦克市,美国)。文章:fRNAkenseq: a fully powered-by-CyVerse cloud integrated RNA-sequencing analysis tool 也是类似,我们团队优秀的NGS组学授课讲师:张娟解读了这篇文章。
关于这个杂志,搜到技能树以前的一个帖子,挺有意思的额:peerJ期刊探索
整体来说就一张图就看完了整个文章,额,我找了半天,文章从头找到尾,我本来以为人家开发一个tools,至少的给一个GUI或者啥的统一接口啥的,结果没有,都是基于别人的网页。不知道是不是我眼瞎没找到==还是说是Agave?
文章流程图:
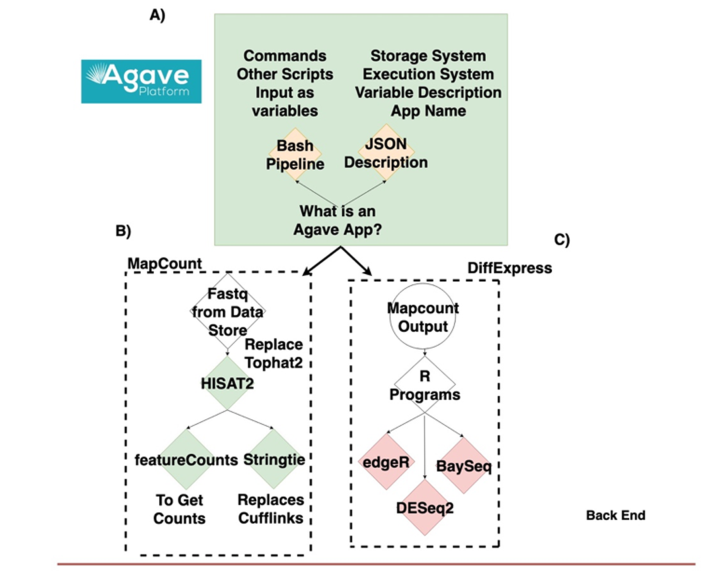
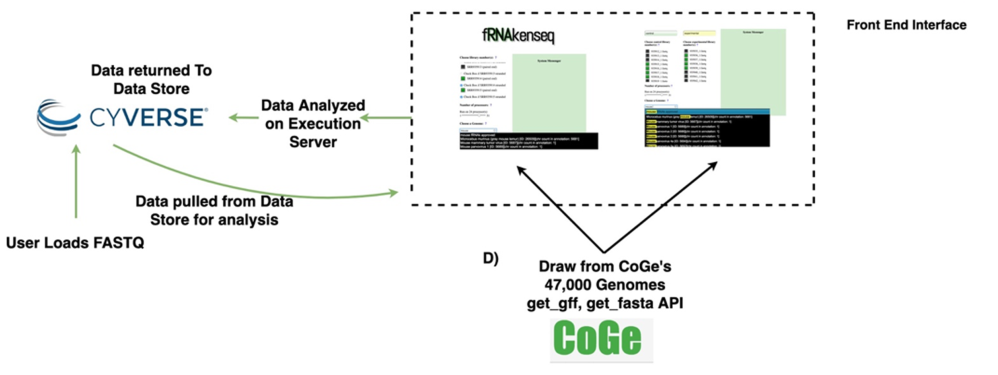
图注:Figure 1 Data lifecycle of fRNAkenseq job and schema illustrating how components across CyVerse and CoGe relate to one another.(A) Outline of Agave App (B) schema for MapCount (C) schema for DiffExpress (D) outline for connectivity with fRNAkenseq and CoGe as well as Data Store. User data uploaded into CyVerse is accessible for analysis by the fRNAkenseq front end. The path followed by user uploaded sequencing data during an analysis is illustrated in the green arrows.使用方法:
1、通过CyVerse Data Store:https://de.cyverse.org/de/ 上传原始fq数据,那么这个地方网速就会成为一个痛点,网站反应速度还行。内嵌形式。后面使用卡顿。体验感不好。
2、通过CoGe(The Comparative Genomics Platform )获得参考基因组fa或者注释文件或者上传自己的参考基因组。
3、A Grid And Visualization Environment (Agave) API system
这个文章中可能比较有意思的地方在于CoGe这个资源。
关于CoGe的介绍:
比较基因组数据库:CoGe: Comparative Genomics https://genomevolution.org/coge/
CoGe is a platform for performing Comparative Genomics research. It provides an open-ended network of interconnected tools to manage, analyze, and visualize next-gen data.
目前该网站首页显示有(2018.03.01)
Organisms: 18,131 Genomes: 33,748 Features: 1,733,099,822 Experiments: 9,467
这是该网站的介绍:FAQs - CoGepedia https://genomevolution.org/wiki/index.php/FAQs#What_is_CoGe.3F
该网站物种量大,数据格式比较规范,容易下载和使用3.1 What is CoGe?
CoGe是一个快速、方便地检索和比较基因组信息和序列数据的在线系统,更新到Aug 1st, 2019,已经有50,000 Genomes in CoGe。
3.2 Why call it CoGe?
CoGe(发音/ kō:jē/)代表比较基因组学(Comparative Genomics)。
3.3 Why make another comparative genomics system?
我们发现,现有的比较基因组系统在其容纳基因组信息的能力上是有限的,为了使其便于比较分析,我们从头开始设计CoGe来解决四个主要的限制:
- 在单一平台上存储来自多个生物的多个基因组的多个版本
- 快速找到感兴趣的基因组中感兴趣的序列(以及相关信息)
- 使用任何算法比较多个基因组区域
- 以这样一种方式可视化分析结果,以便快速和容易地识别“有趣的”模式
总之,我们想要一个比较基因组系统,能让我们尽可能快地测试我们的想法和假设,这样我们就能花更多的时间思考基因组和它们的进化,而不是试图获得和分析基因组序列。
同时,我们意识到,我们需要一个系统,可以让我们快速开发新工具,并添加新的基因组数据,因为它们是可用的。这意味着当我们将一个新的基因组加载到CoGe中,CoGe的所有工具都可以立即进行分析。同样,如果我们开发出一种新工具,用一组基因组解决一个特定问题,那么CoGe中所有的基因组都可以立即使用。3.4 How is CoGe designed and put together?
CoGe的核心设计原则是保持简单和高效。这从底层的计算基础设施扩展到基于web的工具。尽管大规模的比较基因组学分析最好是通过编程和访问CoGe的API来完成,但主要还是基于网络的工具推动了分析。请按照此链接浏览CoGe的系统设计概述。
CoGe’s system design
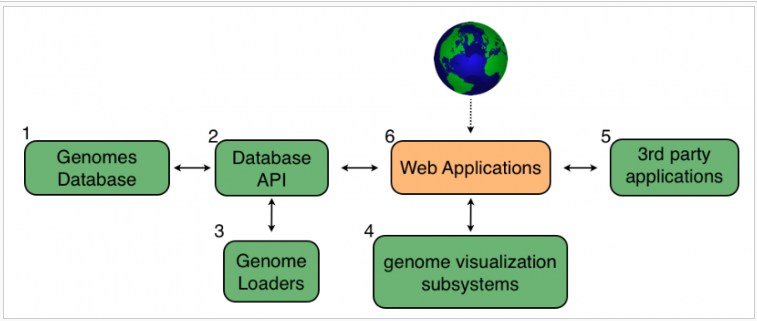
CoGe’s system architecture
CoGe的核心是一个关系数据库(a),用于存储来自多种生物在任何装配状态下的多种版本的基因组。在这个数据库之上是一个应用程序编程接口(API;2)提供对数据库的高级功能访问。使用这个API,几个基因组加载程序(3)搜索各种基因组库,以添加新的基因组到数据库。CoGe利用了几个新的库来可视化各种规模的基因组数据(4)和许多第三方应用程序来进行序列分析(5)。一套基于web的应用程序(6)将这些子系统结合在一起,创建了一套相互关联的软件工具,可为世界各地的研究人员评估
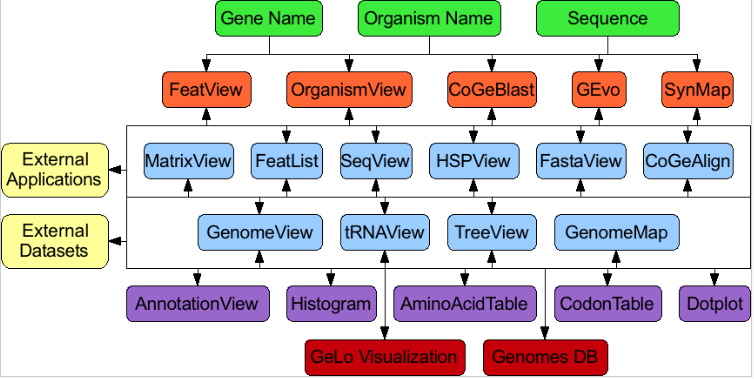
CoGe’s suite of web-based applications creates an open-ended analysis network
这些工具互联的本质(黑线和箭头)创建了一个开放的分析网络,没有预先定义的开始或结束分析。每个工具都专门用于特定类型的数据或分析,都有自己的web应用程序。使用基于web的工具开发框架本质上是为存储分析状态创建了一种方法,这样分析就可以为将来的工作“保存”。这也允许外部工具和数据集容易链接到CoGe为许多类型的数据和分析。绿框是研究人员可以用来开始分析的数据类型。橙色的盒子是允许用户直接访问的应用程序。蓝框是通过另一个工具访问的内部应用程序。紫色盒子是用于显示特定类型数据的网络工具模块。红盒子是CoGe的核心模块。黄色框是CoGe基于web的框架之外的资源。3.5 What is needed to run CoGe?
并不多。只需一个网页浏览器就可以连接到互联网:
- 浏览器设置:推荐浏览器:Chrome或Firefox。其他浏览器如Safari和Internet Explorer也可以工作,但不会定期测试。
- Flash插件
- 启用Javascript(如何在Firefox中启用Javascript)
- 启用弹出窗口(仅供CoGe使用)
- 启用cookie(如果您有一个CoGe用户帐户,这是必要的)
3.6 What is CoGe’s sequence analysis workflow or pipeline?
虽然我们将CoGe设计成易于查找和比较基因组序列,但系统中没有单一的线性工作流程。相反,CoGe的工具创建了一个开放式的分析网络。有一些中心工具和访问点允许您访问系统以查找感兴趣的序列,以及“hub”点将您从系统的一个部分带到另一个部分。这允许在工作时产生想法,并能够迅速扩展到调查任何你发现的有趣现象。当你有了答案,分析就结束了。
例如,您从您最喜爱的基因组(鼠标) - 使用SynMap做一个全基因组的比较它对人类
- 使用SeqView识别一个inversion区域,比较使用各种high-detail断点的区域,提取出人类序列
- 使用CoGeBlast来找到在其它脊椎动物基因组(例如黑猩猩、老鼠和鸭嘴兽)中的同源染色体
- 使用GEvo验证假定的syntenic区域
- 使用FeatView找到一个特定基因额外有趣,因为它在这个syntenic区域的拷贝书变异和然后得到序列
- 使用CoGeBlast找到它的特定内和特定间同源
- 使用FastaView生成这些假定同源的fasta文件,您可以使用CoGeAlign对其进行比对
- 然后使用TreeView构建系统发育树或导出到更广泛的系统发育工具集,如CIPRES。
大量NGS组学等待你发表流程文章哦
B站的不同的数据分析视频课程,见:
- 免费视频课程《RNA-seq数据分析》
- 免费视频课程《WES数据分析》
- 免费视频课程《ChIP-seq数据分析》
- 免费视频课程《ATAC-seq数据分析》
其实如果你也是有时间,愿意耗费时间把这些流程整理成文章,就可以投稿发表哦!