有粉丝提问为什么我免费共享在B站那么多不同的数据分析视频课程,见:
- 免费视频课程《RNA-seq数据分析》交流群组建通知
- 免费视频课程《WES数据分析》交流群组建通知
- 免费视频课程《ChIP-seq数据分析》交流群组建通知
- 免费视频课程《ATAC-seq数据分析》交流群组建通知
有必要学习那么多吗?
这样的问题我只能是笑而不语,选择了科研确实很难走捷径,如果是十年前,当然做一个简单转录组测序就很了不得了,但是科研热点更迭太快,我们现在都不敢保证做几个单细胞转录组就能发表CNS了!
这个时候,多组学联合就非常值得推荐,比如mRNA水平的表达信息和甲基化信号联合,发表在J Cancer. 2019 Oct的文章:Significant Prognostic Values of Differentially Expressed-Aberrantly Methylated Hub Genes in Breast Cancer就是这样的一个例子,其挖掘的表达芯片矩阵和甲基化芯片信号矩阵来源于GEO数据库:
- expression microarray data from GSE54002, GSE65194
- methylation microarray data from GSE20713, GSE32393
各自走差异分析流程,然后组合确定两个基因集:
- 677 upregulated-hypomethylated
- 361 downregulated-hypermethylated genes
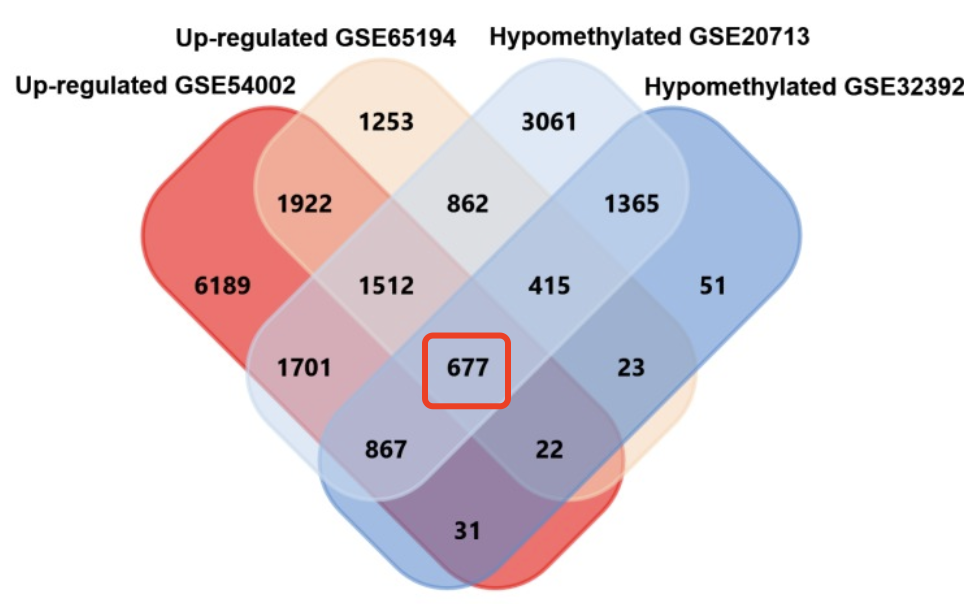
我们提到过数据挖掘的核心是缩小目标基因,比如表达量矩阵通常是2万多个蛋白编码基因,不管是表达芯片还是RNA-seq测序的,采用何种程度的差异分析,最后都还有成百上千个目标基因。如果是临床队列,通常是会跟生存分析进行交集,或者多个数据集差异结果的交集,比如:多个数据集整合神器-RobustRankAggreg包 ,这样的基因集就是100个以内的数量了,但是仍然有缩小的空间,比如lasso等统计学算法,最后搞成10个左右的基因组成signature即可顺利发表。本文比较简单粗暴,直接韦恩图看交集,如下所示:
有了基因集,当然少不了生物学功能数据库的注释,包括GO/KEGG等等。
还可以进行hub基因的策略了,就是去string数据库拿到PPI,然后在cytoscape进行可视化,并且使用插件找hub基因
- 12 hub genes (TOP2A, MAD2L1, FEN1, EPRS, EXO1, MCM4, PTTG1, RRM2, PSMD14, CDKN3, H2AFZ, CCNE2) were sorted from 677 upregulated-hypomethylated genes.
- 4 hub genes (EGFR, FGF2, BCL2, PIK3R1) were sorted from 361 downregulated-hypermethylated genes.
这些分析,基本上读一下我在生信技能树的表达芯片的公共数据库挖掘系列推文 就明白了:
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
当然了,拿到的hub基因通常是会去其它数据库进行验证,还可以生存分析看看它是否可以作为 diagnosis and poor prognosis biomarkers,这样临床意义就升华了。我在生信技能树多次分享过生存分析的细节:
- 基因表达量高低分组的cox和连续变量cox回归计算的HR值差异太大?
- 学徒作业-两个基因突变联合看生存效应
- TCGA数据库里面你的基因生存分析不显著那就TMA吧
- 对“不同数据来源的生存分析比较”的补充说明
- 批量cox生存分析结果也可以火山图可视化
- 既然可以看感兴趣基因的生存情况,当然就可以批量做完全部基因的生存分析
- 多测试几个数据集生存效应应该是可以找到统计学显著的!
- 我不相信kmplot这个网页工具的结果(生存分析免费做)
- 为什么不用TCGA数据库来看感兴趣基因的生存情况
- 200块的代码我的学徒免费送给你,GSVA和生存分析
- 集思广益-生存分析可以随心所欲根据表达量分组吗
- 生存分析时间点问题
- 寻找生存分析的最佳基因表达分组阈值
- apply家族函数和for循环还是有区别的(批量生存分析出图bug)
- TCGA数据库生存分析的网页工具哪家强
- KM生存曲线经logRNA检验后也可以计算HR值
只有掌握多种数据分析技能
才能做到不同数据组合分析,在生命科学领域的科研道路上走的更远!
不过,我这里有一个疑问,这个J Cancer. 2019 Oct的文章为什么要采用不同数据集的mRNA水平的表达信息和甲基化信号联合分析呢?难道TCGA数据库的同一个病人队列的mRNA水平的表达信息和甲基化信号信息不是更优吗?
不同数据集的不同病人群体,混杂的因素太多太多了,至少TCGA数据库是同一个病人队列!