如果大家学过我免费共享在B站上不同的数据分析视频课程,见:
- 免费视频课程《RNA-seq数据分析》交流群组建通知
- 免费视频课程《WES数据分析》交流群组建通知
- 免费视频课程《ChIP-seq数据分析》交流群组建通知
- 免费视频课程《ATAC-seq数据分析》交流群组建通知
可以发现,这些三年前的视频教程里面都是从SRA(Sequence Read Archive)数据库下载文献的测序数据,我也在五年前详细解读过SRA数据库的结构:
层级结构是:SRP(项目)—>SRS(样本)—>SRX(数据产生)—>SRR(数据本身)
伴随数据库是project,层级是PRJNA —> SAMN
链接如下: - https://www.ncbi.nlm.nih.gov/sra?term=SRP078156 查看样本列表
- https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP078156 下载样本ID表格
- https://www.ncbi.nlm.nih.gov/bioproject/PRJNA327548
- https://www.ncbi.nlm.nih.gov/sra?term=SAMN05341212
当然了,实际上是有六种不同的SRA数据库编号,以S开头,官方说明链接:
https://www.ncbi.nlm.nih.gov/books/NBK56913/#search.what_do_the_different_sra_accessi 不过我们不需要掌握那么多。
但是很多学员反馈说,跟着我的代码,下载SRA数据库的文件速度非常感人,也就是十几KB每秒,而我们的测序原始数据经常就几个TB,所以都放弃了。
后来我又统一整理了文献数据下载教程,因为美国的NCBI的SRA与欧洲的EBI-EMBL以及日本的DDBJ数据库共享数据,所以我建议大家去EBI下载,见:使用ebi数据库直接下载fastq测序数据 , 首先使用conda安装asperaconda create -n download conda activate download conda install -y -c hcc aspera-cli conda install -y -c bioconda sra-tools which ascp ## 一定要搞清楚你的软件被conda安装在哪 ls -lh ~/miniconda3/etc/asperaweb_id_dsa.openssh我们已经多次介绍过conda细节了,这里就不再赘述。
- conda管理生信软件一文就够
- 生信技能树B站软件安装视频
- https://www.bilibili.com/video/av28836717
然后就可以使用conda配置好的aspera软件进行高速下载,同时需要学习欧洲的EBI-EMBL以及日本的DDBJ数据库的编号规则:

首先SRA数据库准备放弃存储碱基质量值
但是今天(2020-07-04 )刷朋友圈居然看到了 Heng Li 的推特截图:

居然,SRA数据库准备抛弃用户上传的fastq测序数据里面的质量值。关于fastq格式测序数据
FastQ格式也是序列格式中常见的一种,它存储了生物序列以及相应的质量评价,其序列以及质量信息都是使用一个ASCII字符标示,最初由Sanger开发,目的是将FASTA序列与质量数据放到一起,目前已经成为高通量测序结果的通行标准格式。
FastQ格式和FastA格式都是用来表示序列,其中FastQ格式是4行表示一个序列,而FastA格式只有两行。FastQ格式增加了2行,但其实仅仅是增加了序列对应碱基的质量值信息。
FASTQ文件中每个序列通常有四行: - 1.第一行:必须以“@”开头,后面跟着唯一的序列ID标识符,然后跟着可选的序列描述内容,标识符与描述内容用空格分开;这个与FastA格式的第一行类似,差异就是FastA格式的序列标识通常是大于号”>”开头。
- 2.第二行:序列本身,只允许使用既定的核苷酸或氨基酸编码符号,编码规则见前文的标准IUB / IUPAC 表格;
- 3.第三行:通常就是一个简单加号字符“+”占位即可
- 4.第四行:序列的碱基组成的质量字符,每个字符对应第二行相应位置碱基或氨基酸的质量。
重点是理解质量字符,比如A,首先大家需要理解ASCII码,然后需要理解Phred quality score。
居然还收费
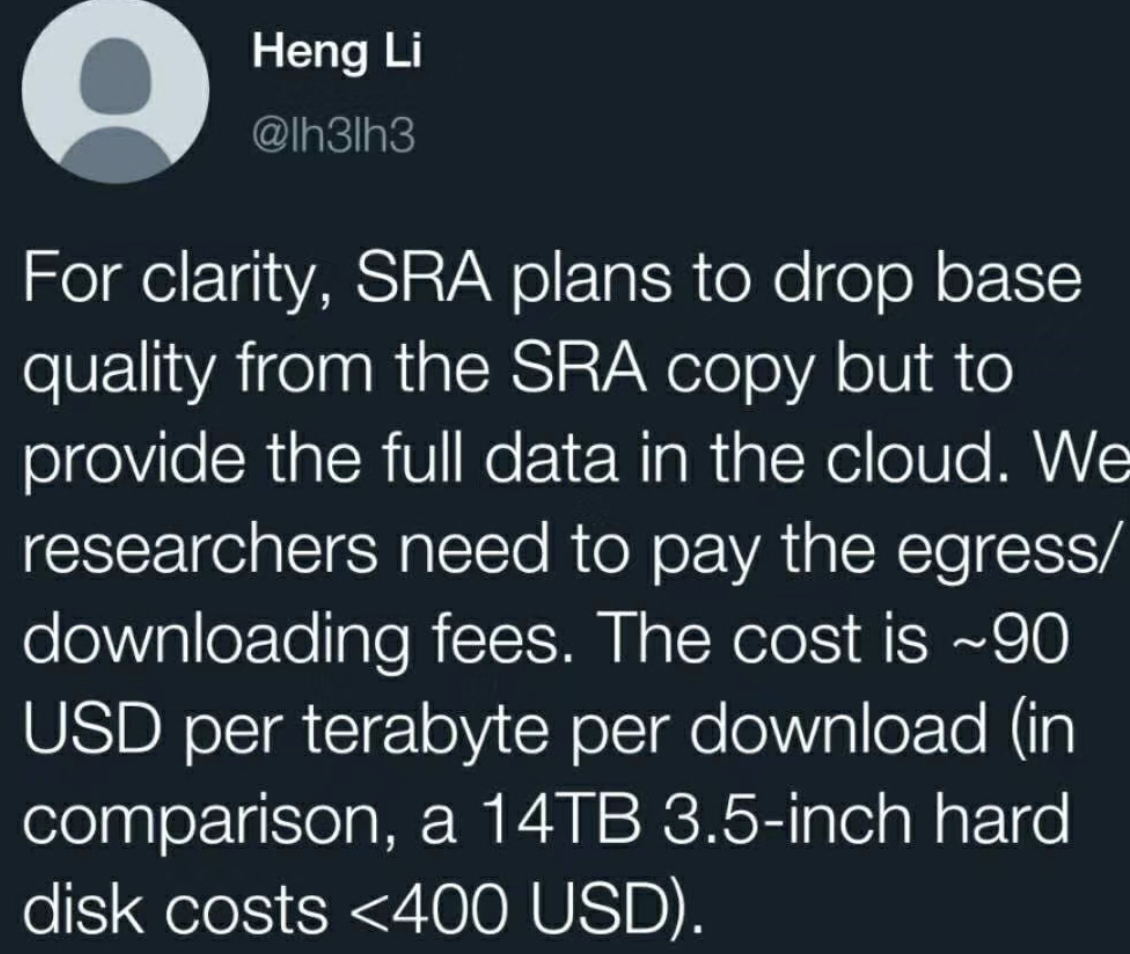
怕粉丝理解不清楚,Heng Li又解释了一番:
不知道是不是受疫情影响?
可以考虑作为国内的测序数据存储中心,大家可以留言推荐一下国内好用的数据库哦!