小白学员好不容易跟完了生信技能树的马拉松式数据挖掘课程,见:
- 数据挖掘学习班第5期(线上直播3周,马拉松式陪伴,带你入门),原价4800的数据挖掘全套课程, 疫情期间半价即可抢购。
- 生信爆款入门-第7期(线上直播4周,马拉松式陪伴,带你入门),原价9600的生信入门全套课程,疫情期间3.3折即可抢购。
现在终于可以开始自己的课题了。
然后····1. 首先是下载数据
gset <- getGEO('GSE16561', destdir=".", AnnotGPL = F, getGPL = F)下载完成后一看数据,
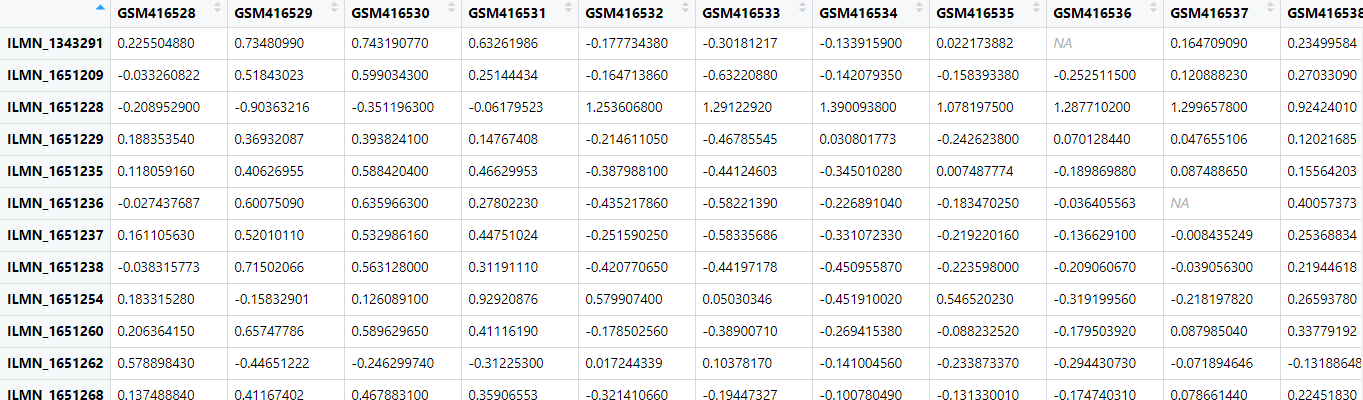
发现好多负值,应该是数据经过背景矫正,log2转换之后又经过scale的数据(z-score的)。也就是小洁老师上课时说的那种不能直接用来做DEG分析的芯片数据。心中顿时飞过一匹草泥马~2. 登录GEO查看原始数据
没办法了,只能是亲自去GEO界面查看该数据集,果然,这个芯片从来没有听说过,应该是很小众了。GPL是Illumina 的 beadchip,GPL号还不在生信技能树大神整理的注释包列表中(http://www.bio-info-trainee.com/1399.html),不过有rio包,注释信息到方便解决,但是**依然隐隐有种前途堪忧的预感。**
不管三七二十一,先把能下下来的写着RAW的东东都下下来解压后检查一遍再说。
然而……让人难过的事情再次发生了….

定睛一看这个raw data的结构,和Jimmy大神的五年前的教程里的数据结构相去甚远(《用lumi包来处理illumina的bead系列表达芯片》 http://www.bio-info-trainee.com/1944.html#more-1944)
另一个raw包里的芯片信息并没有提及rawdata是如何处理的,似乎对后续的数据分析毫无帮助……
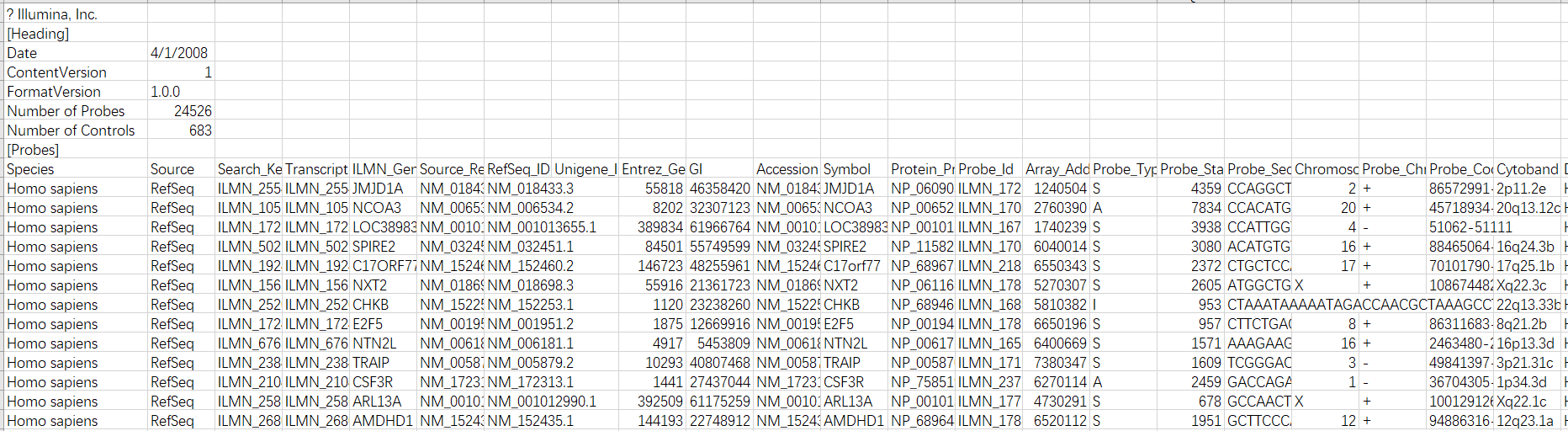
3. 生命不止,折腾不息,查看soft和作者原文
但是,小白我并不想放弃这个GSE,就想看看这个rawdata是怎么处理的,基于生命不止,折腾不息的精神于是我又把soft文件下了下来,定睛一看:
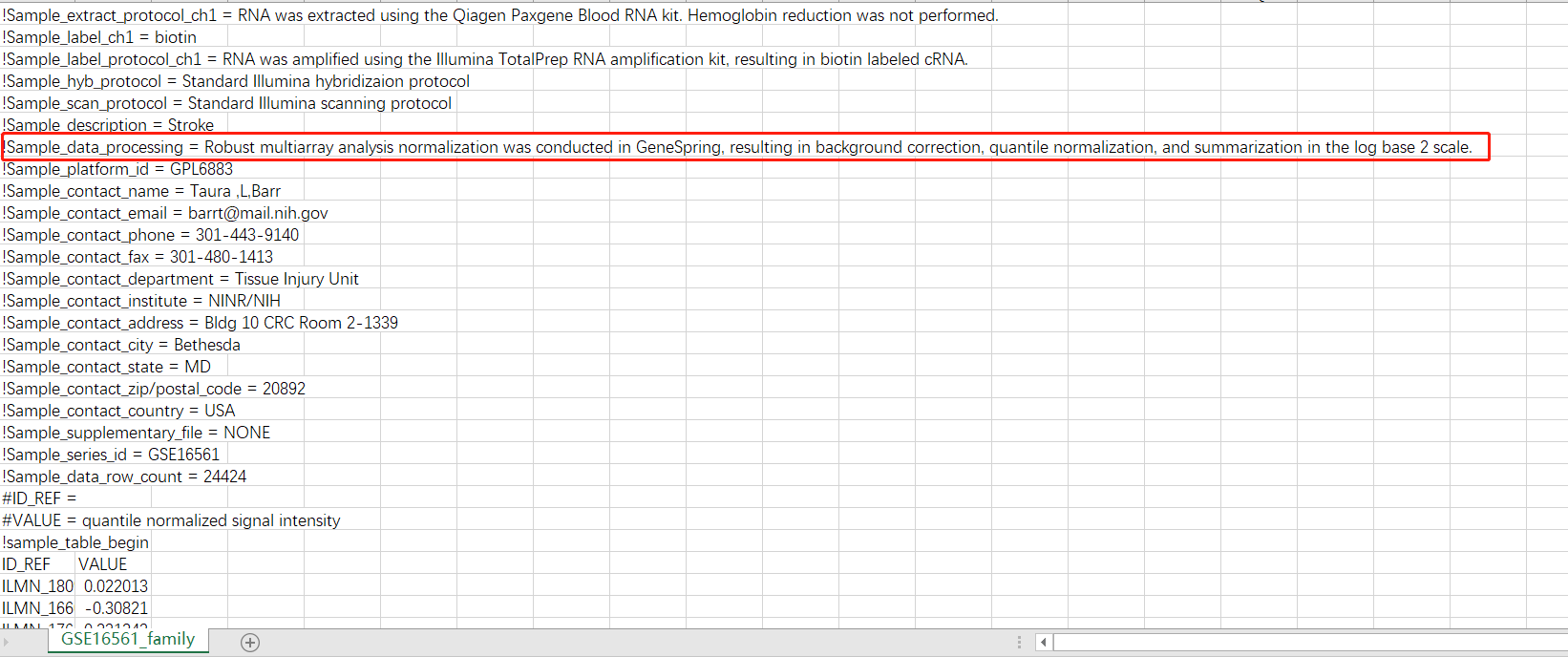
似乎我们离真相又靠近了一点,起码知道这个rawdata大概经历了什么才变成了一开始看到的带了负值的scale数据。此外我还下载了该数据的SCI原文,里面的信息也和我的理解差不多。

4. 大胆假设,小心求证,不服就干,反正错了又不要钱
不过又有几个问题摆在了我的面前:
- 这个GeneSpring软件处理的背景矫正用的都是哪些探针?
- 我们看到的rawdata是经过背景矫正的么?
因为查不到具体的作为背景矫正用的control探针的信息,作为一个初生牛犊不怕虎的生信小白,我做出了一个大胆的决假设:假定我们之前看到的rawdata是经过背景矫正的,反正也是死马当活马医、错了也不要钱,不服就干……
一顿操作猛如虎,经过quantile normalization和log2转换后,我终于画出了一张漂亮的boxplot……
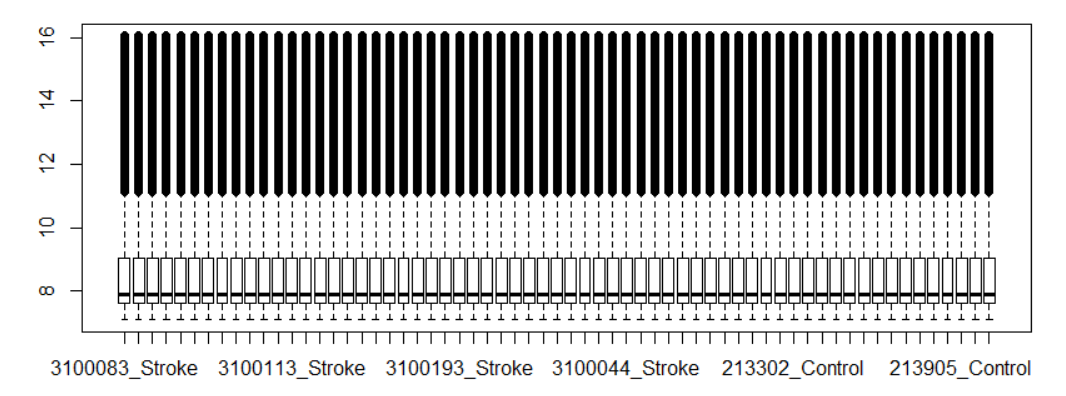
为了证实我的操作是否正确,我把这个图发给了Jimmy大神,在收到Jimmy大神的肯定之后,我欢快的进行了后续的DEG分析,当我满心欢喜的查看分析结果时,悲剧再次发生了,我的差异基因结果如下:
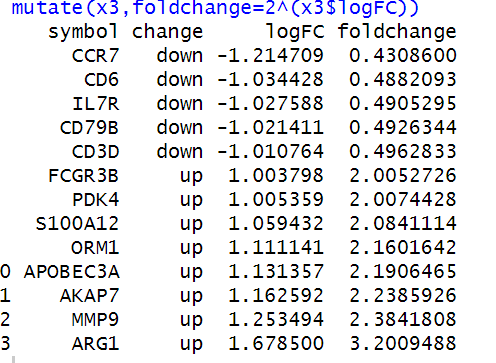
然而原作者的结果如下:
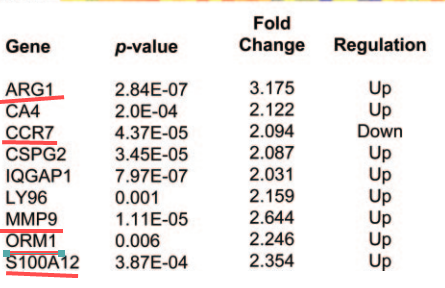
差别还是有些大……怀着忐忑的心情,我再次联系上了Jimmy大神,Jimmy大神尽然爽快的答应帮我看一看这个数据集,简直不敢相信大神肯出手相救,在这里请大神收下我的膝盖!
高效的Jimmy大神很快将他的结果和代码发给我了,大神就是大神,复现的结果和原作者基本相同(其中log的底数是e):
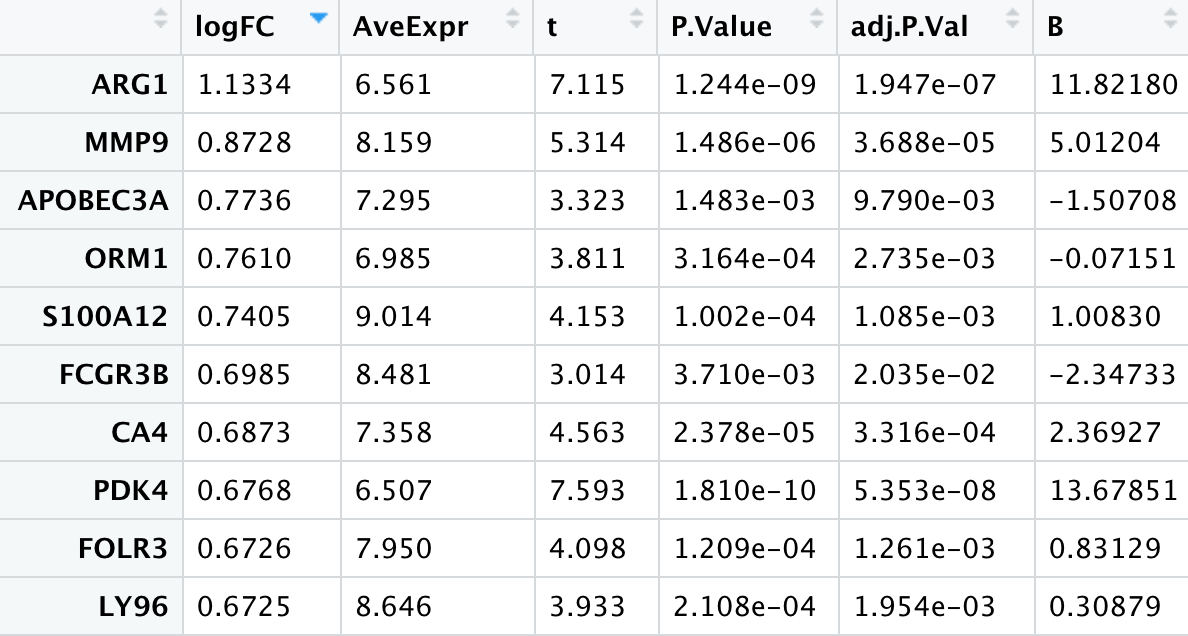
我仔细的拜读了一遍大神的代码,发现原来我只顾着进行quantile normalization和log2转换了,却忽略了小洁老师课里给我们讲的去除重复探针的操作。导致最终的结果与原作者相差甚远。这里附上Jimmy大神的代码(未经大神同意就po出大神的代码,还希望大神不要见怪):
```R
b=read.table(‘GSE16561RAW.txt’,header = T)
b[1:4,1:4]
rownames(b)=b[,1]
b=b[,-1]
b=log(b+1)
boxplot(b,las=2)
dat=b
library(limma)
dat=normalizeBetweenArrays(dat)
boxplot(dat,las=2)
pd=pData(a)
colnames(dat)
library(stringr)
group_list= str_split(colnames(dat) ,’‘,simplify = T)[,2]
table(group_list)
dat[1:4,1:4]
a这个 AnnoProbe 包是jimmy大神为了拯救我们小白亲自开发的!
library(AnnoProbe)
ids=idmap(‘GPL6883’,type = ‘soft’)
head(ids)
colnames(ids)=c(‘probe_id’,’symbol’)
ids=ids[idsprobe_id %in% rownames(dat),]
dat[1:4,1:4]
dat=dat[idsmedian=apply(dat,1,median) #ids新建median这一列,列名为median,同时对dat这个矩阵按行操作,取每一行的中位数,将结果给到median这一列的每一行
ids=ids[order(idsmedian,decreasing = T),]
对ids
median中位数从大到小排列的顺序排序,将对应的行赋值为一个新的ids
ids=ids[!duplicated(ids
probe_id,] #新的ids取出probe_id这一列,将dat按照取出的这一列中的每一行组成一个新的dat
rownames(dat)=ids$symbol#把ids的symbol这一列中的每一行给dat作为dat的行名
dat[1:4,1:4] #保留每个基因ID第一次出现的信息(median最大的那个)
save(dat,group_list,file = ‘step1-output.Rdata’)
```
经过大神的代码处理之后的rawdata再拿来跑一遍差异分析的流程,后续结果就令人十分满意了!
5. 小结
- 小白这次大胆的探索真的是歪打正着,在这里写下手账,希望可以给那些被类似数据困扰的小伙伴们提供一定的参考。
- 做rawdata处理的时候一定不要忘记去重!不要忘记去重!不要忘记去重!。
6. 致谢
本人临床背景,生信零基础,本着对生信的一腔热情,联系上了Jimmy大神,参加了生信技能树的学习班!特别感谢Jimmy大神的启发式引导、还有小洁老师精彩的R语言课程和时不时给我灌下的毒鸡汤!
因为个人的工作时间的调整问题,后半程郭老师和张老师的linux课程我大多数时候无法参加线上直播,但好在有回放可以看,这里要给两位老师说一声抱歉,因为不能参与到直播互动中来而浪费了你们精心备好的课!
总之,很荣幸能够找到生信技能树,向各位老师讨教学习,这真的是一段很棒的经历。
by:Jack Sparrow, 一个生信零基础的小白学员。
2020/08/01文末友情推荐
要想真正入门生物信息学建议务必购买全套书籍,一点一滴攻克计算机基础知识,书单在:什么,生信入门全套书籍仅需160 。
如果大家没有时间自行慢慢摸索着学习,可以考虑我们生信技能树官方举办的学习班:
- 数据挖掘学习班第5期(线上直播3周,马拉松式陪伴,带你入门),原价4800的数据挖掘全套课程, 疫情期间半价即可抢购。
- 生信爆款入门-第7期(线上直播4周,马拉松式陪伴,带你入门),原价9600的生信入门全套课程,疫情期间3.3折即可抢购。
如果你课题涉及到转录组,欢迎添加一对一客服:详见:你还在花三五万做一个单细胞转录组吗?
号外:生信技能树知识整理实习生招募,长期招募,也可以简单参与软件测评笔记撰写,开启你的分享人生!另外:绝大部分生信技能树粉丝都没有机会加我微信,已经多次满了5000好友,所以我开通了一个微信好友,前100名添加我,仅需150元即可,3折优惠期机会不容错过哈。我的微信小号二维码在:0元,10小时教学视频直播《跟着百度李彦宏学习肿瘤基因组测序数据分析》