最近接到粉丝求助,他最近在跟着我B站课程和GitHub代码处理GEO 芯片:
GSE113486 GEO平台 已经进行了log2 转换和 Normalized signal intensity by internal control miRNAs. 我画了他们的箱式图是这样的。不太整齐。我不确定是不是还要进行normalizeBetweenArrays处理。我看你的视频课程是说要样本间要整齐,但是课题组师兄师姐说他们没看这个,直接做差异分析的,所以比较困惑后续处理的细节把握。
箱线图如下:
可以看到,肿瘤样品的表达量整体就比正常对照样品的表达量高出一大截,这样的数据进行后续分析,就会出现大量的上调基因。
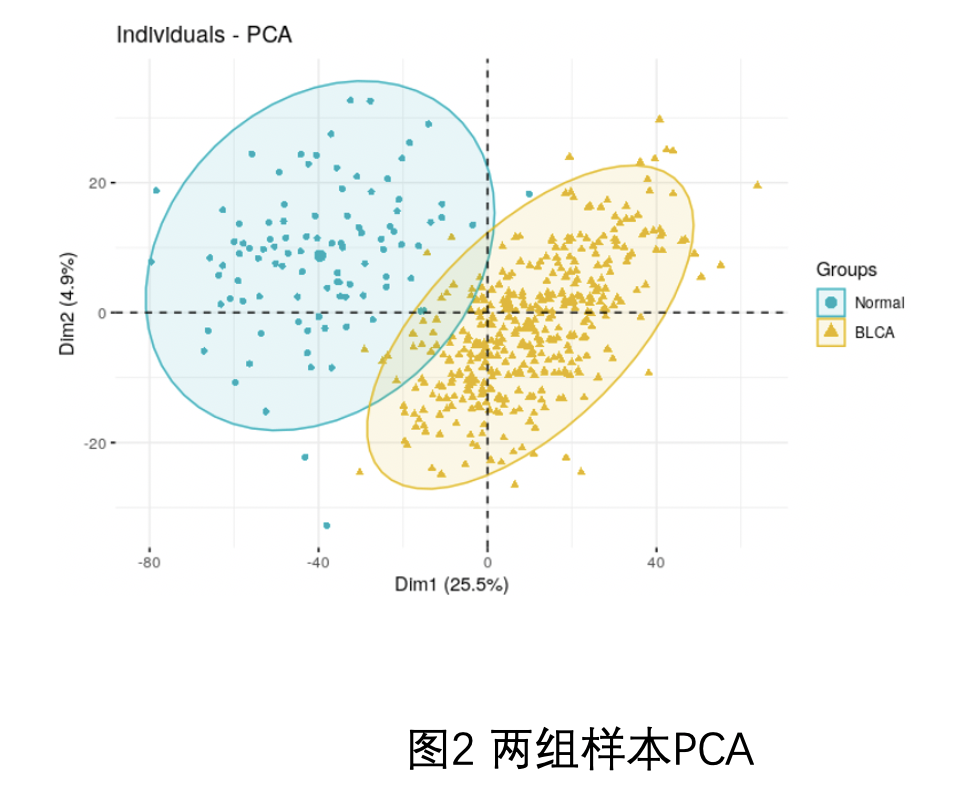
因为我一直强调,做表达矩阵分析一定要有三张图,见:你确定你的差异基因找对了吗? ,所以就让粉丝继续摸索,其中PCA如下:
可以看到,两个分组是泾渭分明的,这可能是生物学差异,因为肿瘤样品就肯定跟正常组织不一样的啊,也有可能是批次效应。所以我给粉丝的建议是两个策略
- 第一个策略是直接normalizeBetweenArrays处理,然后走差异分析。
- 第二是先去除批次效应,然后走差异分析。
建议你比较一下,这两个差异分析的区别。
然后粉丝的行动也很迅速,两三天就回复了邮件,给出了两个摸索结果:
可以看到,直接normalizeBetweenArrays处理其实就是一个quantile的normalization而已,大家可以去看quantile normalization到底对数据做了什么 - 简书,了解一下。
从韦恩图可以看到,没有进行normalizeBetweenArrays处理之前呢,上调基因真的是超级多啊!经过了normalizeBetweenArrays处理之后呢,其中616个上调基因变成了没有显著性改变的基因,然后637个居然由上调基因变成了下调基因,当然了,也有341个基因维持原来的上调属性。
是不是很可怕!!!
粉丝下的这个结论很正确,这个时候使用 limma 的 removeBatchEffect 函数来矫正批次效应,肯定是错的,因为完全没有搞清楚矫正批次效应的统计学原理。
其实几年前我在《单细胞天地》公众号发起过一个谈论,见:到底是批次效应还是真实生物学差异,如果你仅仅是做了两个单细胞转录组样品,想合并这两个数据再后续分析,就面临着两个样品(处理前后的生物学差异)本身的批次效应(不同时间点取样,不同10x上机时间等等)。因为是单细胞,一个样品里面本身就有这成千上万个细胞,可以针对两个样品内部的某些具有不变属性的单细胞来作为锚定,从而比较好的合并两个样品的单细胞转录组数据。
但是,如果是bulk转录组测序,或者表达量芯片,就基本上不可能做到区分具有生物学差异的两个样品的批次效应了。虽然说我在《生信技能树》写过不少相关教程,比如:多种批次效应去除的方法比较,但那样的去除是针对生物学差异与批次效应交叉的情况来去除。比如:- 第一个批次:2个处理,2个对照样品
- 第二个批次:3个处理,3个对照样品
这个时候,就可以使用 limma 的 removeBatchEffect 函数或者 sva 的 ComBat 函数,把批次效应去除掉,然后保留生物学差异供后续的差异分析。
但是如果你的实验设计是:- 第一个批次:3个处理样品
- 第二个批次:3个对照样品
那我就只能奉劝你,对这个数据集说拜拜了!文末友情推荐
要想真正入门生物信息学建议务必购买全套书籍,一点一滴攻克计算机基础知识,书单在:什么,生信入门全套书籍仅需160 。
如果大家没有时间自行慢慢摸索着学习,可以考虑我们生信技能树官方举办的学习班:- 数据挖掘学习班第7期(线上直播3周,马拉松式陪伴,带你入门),原价4800的数据挖掘全套课程, 疫情期间半价即可抢购。
- 生信爆款入门-第9期(线上直播4周,马拉松式陪伴,带你入门),原价9600的生信入门全套课程,疫情期间3.3折即可抢购。
如果你课题涉及到转录组,欢迎添加一对一客服:详见:你还在花三五万做一个单细胞转录组吗?
号外:生信技能树知识整理实习生招募,长期招募,也可以简单参与软件测评笔记撰写,开启你的分享人生!另外,:绝大部分生信技能树粉丝都没有机会加我微信,已经多次满了5000好友,所以我开通了一个微信好友,前100名添加我,仅需150元即可,3折优惠期机会不容错过哈。我的微信小号二维码在:0元,10小时教学视频直播《跟着百度李彦宏学习肿瘤基因组测序数据分析》