我们的CNS图表复现之旅已经开始,前面3讲是;
如果你也想加入交流群,自己去:你要的rmarkdown文献图表复现全套代码来了(单细胞)找到我们的拉群小助手哈。
既让我们根据Cells were defined as non-immune if belonging to a cluster low for PTPRC (gene for CD45)的规则,把细胞区分为了免疫细胞和非免疫细胞,详见:我在单细胞天地的教程:是否是免疫细胞很容易区分那是否是肿瘤细胞呢?
现在我们就可以来复现文章里面的免疫细胞亚群再分类了,如下:

这幅图超级容易理解,就是13431个免疫细胞,可以继续细分为不同的亚群,如上所述。
首先我们挑选免疫单细胞
因为这个是系列连载教程,所以务必看前面的前面3讲是;
知道我是把全部的2万多个细胞的表达矩阵读入R,变成了Seurat对象,然后判断了细胞是否属于免疫细胞哈。前面的代码核心步骤是:
sce@meta.data$immune_annotation <-ifelse(sce@meta.data$RNA_snn_res.0.5 %in% imm ,'immune','non-immune')
# MAke a table
table(sce@meta.data$immune_annotation)
phe=sce@meta.data
save(phe,file = 'phe-of-immune-or-not.Rdata')
就是把细胞是否属于免疫细胞这个信息(phe=sce@meta.data)保存下来了。后面就可以直接使用啦:
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
library(ggplot2)
load(file = 'first_sce.Rdata')
sce <- FindClusters(sce, resolution = 0.5)
table(sce@meta.data$RNA_snn_res.0.5)
load(file = 'phe-of-immune-or-not.Rdata')
table(phe$immune_annotation)
cells.use <- row.names(sce@meta.data)[which(phe$immune_annotation=='immune')]
length(cells.use)
sce <-subset(sce, cells=cells.use)
sce
继续走Seurat标准流程之聚类分群
这样就拿到了免疫细胞单独进行后续分析,仍然是走Seurat标准流程之聚类分群,代码是:
sce
sce <- NormalizeData(sce, normalization.method = "LogNormalize",
scale.factor = 10000)
GetAssay(sce,assay = "RNA")
sce <- FindVariableFeatures(sce,
selection.method = "vst", nfeatures = 2000)
sce <- ScaleData(sce)
sce <- RunPCA(object = sce, pc.genes = VariableFeatures(sce))
res.used <- 0.7
sce <- FindClusters(object = sce, verbose = T, resolution = res.used)
set.seed(123)
sce <- RunTSNE(object = sce, dims = 1:15, do.fast = TRUE)
DimPlot(sce,reduction = "tsne",label=T)
DimPlot(sce,reduction = "tsne",label=T, group.by = "patient_id")
table(sce@meta.data$seurat_clusters)
然后走singleR的自动注释步骤:
sce_for_SingleR <- GetAssayData(sce, slot="data")
sce_for_SingleR
library(SingleR)
hpca.se <- HumanPrimaryCellAtlasData()
hpca.se
clusters=sce@meta.data$seurat_clusters
pred.hesc <- SingleR(test = sce_for_SingleR, ref = hpca.se, labels = hpca.se$label.main,
method = "cluster", clusters = clusters,
assay.type.test = "logcounts", assay.type.ref = "logcounts")
table(pred.hesc$labels)
celltype = data.frame(ClusterID=rownames(pred.hesc), celltype=pred.hesc$labels, stringsAsFactors = F)
sce@meta.data$singleR=celltype[match(clusters,celltype$ClusterID),'celltype']
DimPlot(sce, reduction = "tsne", group.by = "singleR")
phe=sce@meta.data
table(phe$singleR)
save(phe,file = 'phe-of-subtypes-Immune-by-singleR.Rdata')
分群结果的对比
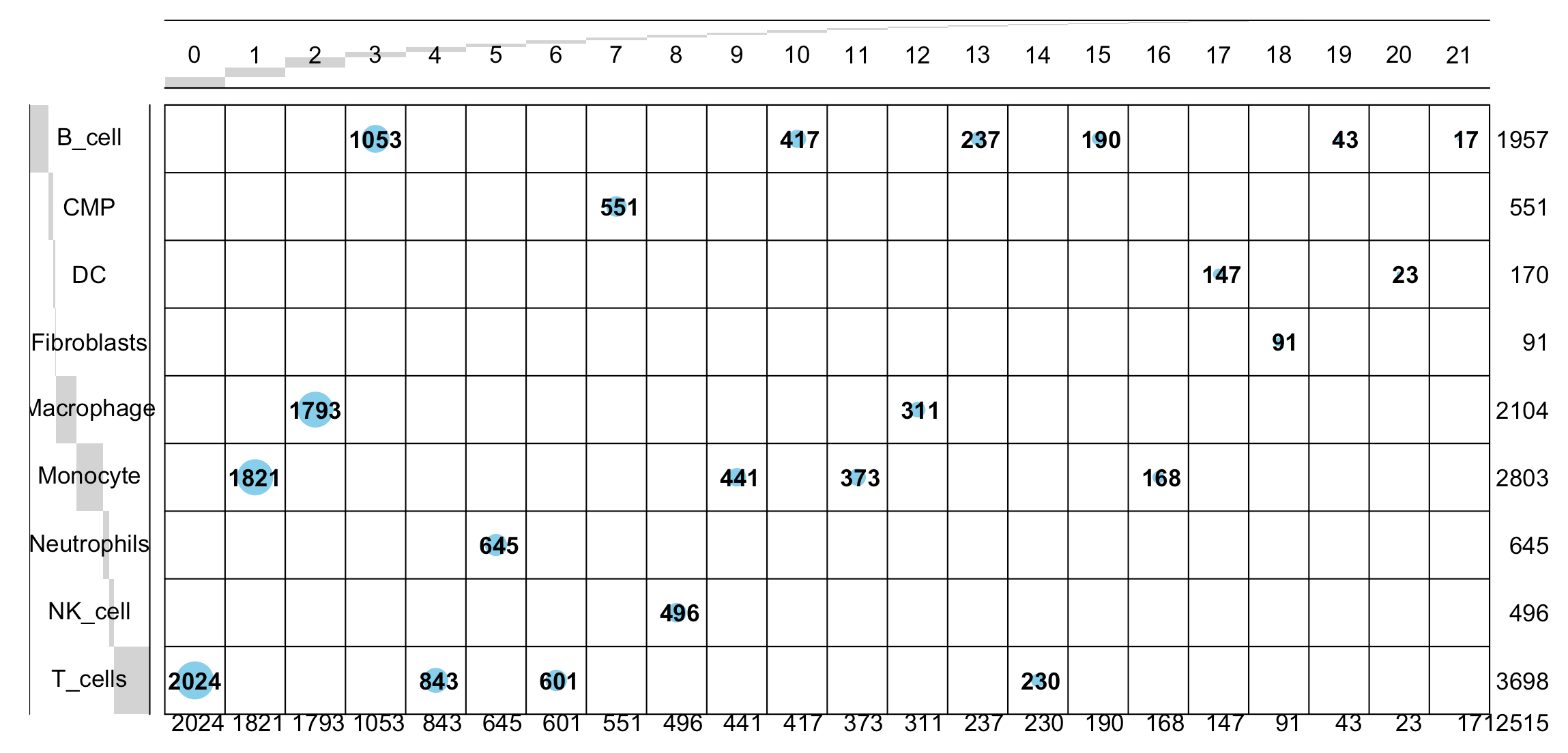
但是我们的这个距离分群结果,跟文章是有一定差异的,如下:
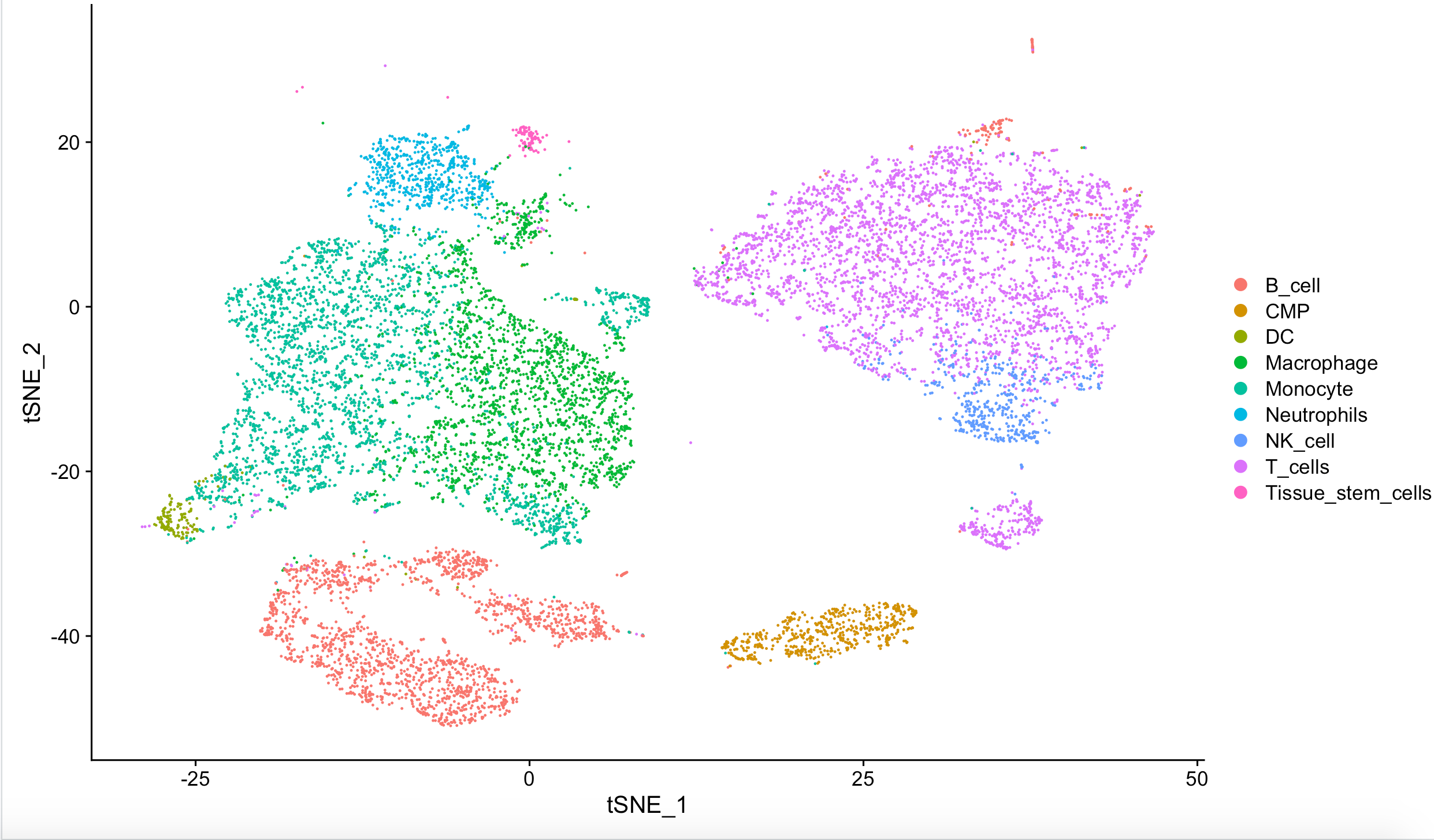
分群是:
> as.data.frame(sort(table(phe$singleR)))
Var1 Freq
1 Fibroblasts 91
2 DC 170
3 NK_cell 496
4 CMP 551
5 Neutrophils 645
6 B_cell 1957
7 Macrophage 2104
8 Monocyte 2803
9 T_cells 3698
但是它们仍然是可以细分的,如下:
文章是:

简单总结一下,文章把Macrophage和Monocyte合并起来,把B_cell区分成为两群。而我们的T_cells和NK_cell也需要被合并起来。而且 common myeloid progenitor (CMP) 这群细胞呢,在文章并没有出现,不知道是不是会被命名为 Mast-cells,需要相应的生物学背景。
文章给出的代码是;
# Annotate each of the clusters
free_annotation <- c("T-cells","MF-Monocytes", "MF-Monocytes", "B-cells-PB", "MF-Monocytes", "T-cells", "T-cells", "Neutrophils", "Dendritic", "Mast-cells", "MF-Monocytes", "T-cells", "B-cells-M", "Unknown", "T-cells", "pDCs", "B-cells-M", "MF-Monocytes")
# free_annotation <- c("0","1", "2", "3", "4", "5", "6", "7", "8", "9", "10", "11", "12", "13", "14", "15", "16", "17", "18")
但是我们中间很多步骤都没有精准的模仿文章的各个流程和参数,所以这个时候的差异就没办法解决了,我们是22个群,文章是19个群。
文末友情推荐
要想真正入门生物信息学建议务必购买全套书籍,一点一滴攻克计算机基础知识,书单在:什么,生信入门全套书籍仅需160 。
如果大家没有时间自行慢慢摸索着学习,可以考虑我们生信技能树官方举办的学习班:
- 数据挖掘学习班第7期(线上直播3周,马拉松式陪伴,带你入门),原价4800的数据挖掘全套课程, 疫情期间半价即可抢购。
- 生信爆款入门-第9期(线上直播4周,马拉松式陪伴,带你入门),原价9600的生信入门全套课程,疫情期间3.3折即可抢购。
如果你课题涉及到转录组,欢迎添加一对一客服:详见:你还在花三五万做一个单细胞转录组吗?
号外:生信技能树知识整理实习生招募,长期招募,也可以简单参与软件测评笔记撰写,开启你的分享人生!另外,:绝大部分生信技能树粉丝都没有机会加我微信,已经多次满了5000好友,所以我开通了一个微信好友,前100名添加我,仅需150元即可,3折优惠期机会不容错过哈。我的微信小号二维码在:0元,10小时教学视频直播《跟着百度李彦宏学习肿瘤基因组测序数据分析》