首发于生信技能树的公众号:人人都可以学会WGCNA(学徒数据挖掘)
我在⽣信技能树写了一系列WGCNA教程,见:
- ⼀⽂看懂WGCNA 分析(2019更新版) (点击阅读原⽂即可拿到示例数据)
- 通过WGCNA作者的测试数据来学习
- 重复⼀篇WGCNA分析的⽂章(代码版)
- 重复⼀篇WGCNA分析的⽂章(解读版)(逆向收费读⽂献2019-19)
- 关键问题答疑:WGCNA的输⼊矩阵到底是什么格式
大量的学员follow下来了我的教程,并且完成了自己的图表复现:
接下来要分享的也是一位优秀学徒的笔记:
学习WGCNA第一步:复现文献
- 题目:Coexpression modules constructed by weighted gene co‑expression network analysis indicate ubiquitin‑mediated proteolysis as a potential biomarker of uveal melanoma
- 数据集:GSE44295
- 平台:GPL6883
- https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE44295
什么是WGCNA?
-
其全称为weighted correlation network analysis, 直译就是加权基因相关性网络分析。通过这项分析,可以鉴定出相关性非常高的基因,即拥有相似的功能的集合,称之为modules,而且可以将modules与表型数据进行关联分析,挖掘潜在的关键基因。
-
分析步骤:
- 构建共表达网络
- 模块识别
- 模块与表型信息关联
- 模块间相关性
- 找出关键基因进行后续验证
- 一句话总结:从共表达网络角度,在大量基因中挖掘出几个具有生物学意义的hub基因。
原文思路
-
数据下载:GSE44295,筛选出其中58个葡萄膜黑色素瘤(UM)样本信息。
-
基因注释:GPL6883,多个探针对应下取最大平均值;有表达量负值的基因删掉。
- WGCNA分析:取平均值前5000个基因进行分析
- GO,KEGG通路富集分析:结果显示模块2主要富集与转录调控相关的通路中,且模块2-4均在泛素介导的蛋白水解途径中富集
- 结论:
- 模块2被认为是UM发生的关键模块
- 泛素介导的蛋白水解途径可能在UM发生发展中发挥重要作用

- 因为是刚开始学习WGCNA,所以先给自己立两个目标
- 先过一遍代码
- 学习看每张图,看如何写结论
第一步:数据下载
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)#在调用as.data.frame的时,将stringsAsFactors设置为FALSE可以避免character类型自动转化为factor类型
# 注意查看下载文件的大小,检查数据
##网速不好,用geochina
#library(devtools)
#library(GEOmirror)
geoChina('GSE44295')
load('GSE44295_eSet.Rdata') ## 载入数据
class(gset) #查看数据类型
length(gset) #
class(gset[[1]])
gset
# assayData: 24526 features, 63 samples
# 因为这个GEO数据集只有一个GPL平台,所以下载到的是一个含有一个元素的list
a=gset[[1]] #
exprSet=exprs(a)
dim(exprSet)
head(exprSet[,1:4])
boxplot(exprSet,las=2)
exprSet
dat=exprSet
#提取临床信息
pdata<-pData(a)
#筛选样本为58个
table(pdata$source_name_ch1)
which(pdata$source_name_ch1=='primary uveal melanoma cells')
pdata=pdata[which(pdata$source_name_ch1=='primary uveal melanoma cells'),]
第二步:基因注释
###基因注释
#https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL6883
#从Platforms GPL6883下载的注释文件,改名为ann.txt
ann=read.delim(file='GPL6883.txt',comment.char = '#',stringsAsFactors = F)
colnames(ann)
dat=as.data.frame(dat)
#将筛选样本,与临床数据对应
dat=dat[,rownames(pdata)]
dat$ID=rownames(dat)
#与注释信息整合在一起
id_symbol_expr<-na.omit(merge(x=dat,y=ann[c('ID','Symbol')],by='ID',all.x=T))
#这里处理一个探针对应不同gene名的情况,随便取一个就行
symbol<-lapply(id_symbol_expr$Symbol,FUN = function(x){strsplit(x,'///')[[1]][1]})
id_symbol_expr$Symbol<-as.character(symbol)
#去除掉NA值,就是说有些探针对应不到已知的基因上,至少在GPL文件中没有对应关系
ids=id_symbol_expr[id_symbol_expr$Symbol != 'NA',]
ids=ids[ids$ID %in% rownames(dat),]
dat=dat[ids$ID,]
#去掉最后一列ID
dat=dat[,-59]
#处理一个gene名对应多个探针情况,平均数排序取最大
ids$mean=apply(dat,1,mean) #ids新建mean这一列,列名为mean,同时对dat这个矩阵按行操作,取每一行的平均数,将结果给到mean这一列的每一行
ids=ids[order(ids$Symbol,ids$mean,decreasing = T),]#对ids$symbol按照ids$mean从大到小排列的顺序排序,将对应的行赋值为一个新的ids
ids=ids[!duplicated(ids$Symbol),]#将symbol这一列取取出重复项,'!'为否,即取出不重复的项,去除重复的gene ,保留每个基因最大表达量结果s
dat=dat[ids$ID,] #新的ids取出probe_id这一列,将dat按照取出的这一列中的每一行组成一个新的dat
rownames(dat)=ids$Symbol#把ids的symbol这一列中的每一行给dat作为dat的行名
dat[1:4,1:4] #保留每个基因ID第一次出现的信息
save(dat,ann,pdata, file = "step1.Rdata")
第三步:聚类树形图
##-----------------------------------------------------------------按照文献要求处理数据
#将有负值的样本删掉 保存成dat
for (i in 1:58) {
dat=dat[which(dat[,i]>0),]
}
dim(dat) #7469 58
#计算平均值,取前5000个
dat$mean=apply(dat,1,mean)
dat=dat[order(dat$mean,decreasing = TRUE),]
five=dat[1:5000,]
save(five,file='five.Rdata')
##聚类树形图
five=t(five)
five=as.data.frame(five)
five=five[-59,]
sampleTree = hclust(dist(five), method = "average");
plclust(sampleTree)
datExpr_tree <- hclust(dist(five), method = "average")
par(mar = c(0,5,2,0))
plot(datExpr_tree, main = "Sample clustering", sub = "", xlab = "",
cex.axis = 0.9, cex.main = 1.2, cex.lab = 1, cex = 0.7)


第四步:确定软阈值
rm(list=ls())
# Load the WGCNA package
library(WGCNA)
# The following setting is important, do not omit.
options(stringsAsFactors = FALSE);
# Load the data saved in the first part
load(file = 'five.Rdata');
five=t(five)
five=as.data.frame(five)
five=five[-59,]
#=====================================================================================
#
# Code chunk 2
#
#=====================================================================================
#计算软阈值
# Choose a set of soft-thresholding powers
powers = c(c(1:10), seq(from = 12, to=20, by=2))
# Call the network topology analysis function
sft = pickSoftThreshold(five, powerVector = powers, verbose = 5)
# Plot the results:
sizeGrWindow(9, 5)
par(mfrow = c(1,2));
cex1 = 0.9;
# Scale-free topology fit index as a function of the soft-thresholding power
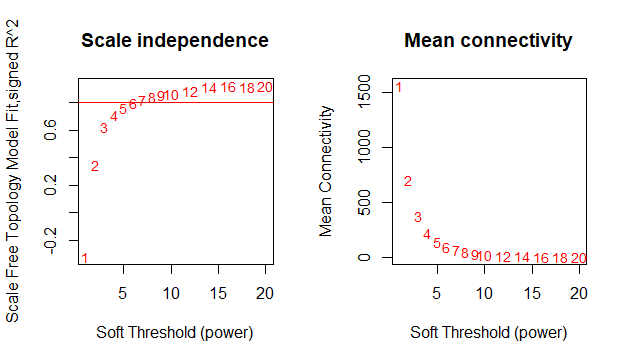
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n",main = paste("Scale independence"));
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],labels=powers,cex=cex1,col="red");
# this line corresponds to using an R^2 cut-off of h
abline(h=0.80,col="red")
# Mean connectivity as a function of the soft-thresholding power
plot(sft$fitIndices[,1], sft$fitIndices[,5],xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col="red")
#软阈值为6

第五步:构建共表达矩阵
#一步构建网络
#报错:https://blog.csdn.net/liyunfan00/article/details/91686840
cor <- WGCNA::cor
#修改power值,最小模块基因数50(按原文)
net = blockwiseModules(five, power = 6,
TOMType = "unsigned", minModuleSize = 50,
reassignThreshold = 0, mergeCutHeight = 0.20,
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs = TRUE,saveTOMFileBase = "femaleMouseTOM",
verbose = 3)
# 改回去
cor<-stats::cor
class(net)
names(net)
table(net$colors)
#0 1 2 3 4 5 6 7
#365 2730 681 379 370 239 176 60
#一共聚类出7个模块,0对应没有聚类到的基因数
save(net,file='net.Rdata')
#=====================================================================================
#
# Code chunk 4
#
#=====================================================================================
moduleColors <- labels2colors(net$colors)
# Recalculate MEs with color labels
MEs0 = moduleEigengenes(five, moduleColors)$eigengenes
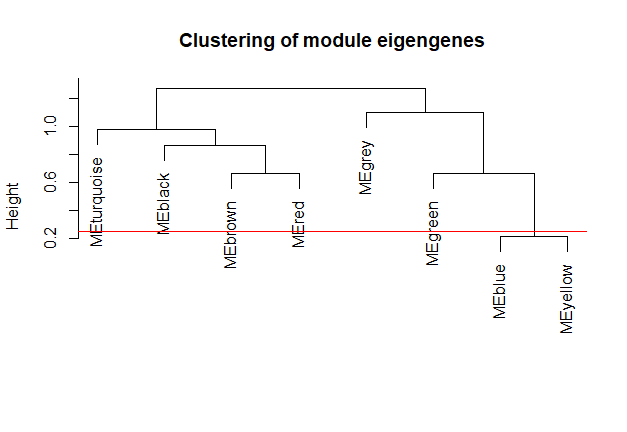
# 计算根据模块特征向量基因计算模块相异度:
MEDiss = 1 - cor(MEs0);
# Cluster module eigengenes
METree = hclust(as.dist(MEDiss), method = "average");
plot(METree,
main = "Clustering of module eigengenes",
xlab = "",
sub = "")
# 在聚类图中画出剪切线
MEDissThres = 0.25
abline(h = MEDissThres, col = "red")
merge_modules = mergeCloseModules(five, moduleColors, cutHeight = MEDissThres, verbose
= 3)
#=====================================================================================
#
# Code chunk 5
#
#=====================================================================================
mergedColors = merge_modules$colors;
mergedMEs = merge_modules$newMEs;
plotDendroAndColors(net$dendrograms[[1]], cbind(moduleColors, mergedColors),
c("Dynamic Tree Cut", "Merged dynamic"),
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
- 我这里用WGCNA把基因分成7个模块,基因数分别是2730 681 379 370 239 176 60,而原文中:

- 与模块大小相关的参数主要是blockwiseModules函数里面的minModuleSize、mergeCutHeight这两个参数。
- 如果这两个参数越小,模块的大小也会越小,模块数量就会增多。
- 而作者文中规定minModuleSize为50,所以应该是mergeCutHeight参数不一致导致结果出现偏差。
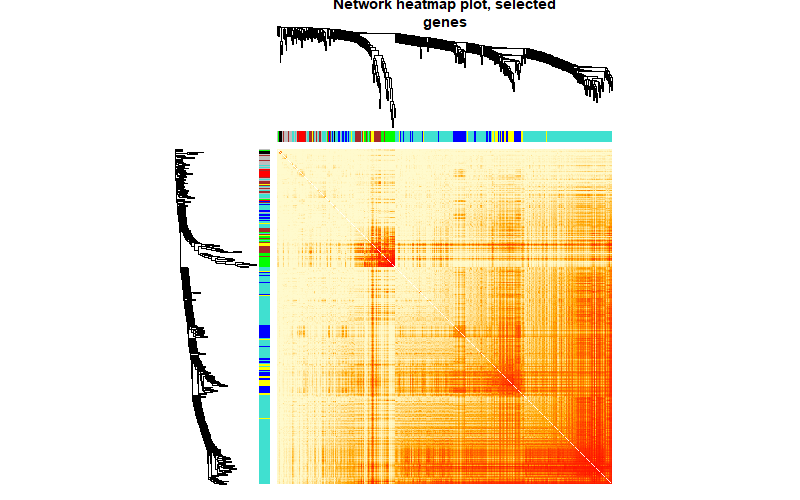
第六步:绘制TOM热图
##绘制TOM热图
# tom plot
nGenes = ncol(five)
nSamples = nrow(five)
geneTree = net$dendrograms[[1]]
dissTOM = 1 - TOMsimilarityFromExpr(five, power = 6)
nSelect = 400
set.seed(10)
select = sample(nGenes, size = nSelect)
selectTOM = dissTOM[select, select]
selectTree = hclust(as.dist(selectTOM), method = "average")
selectColors = moduleColors[select]
sizeGrWindow(9,9)
plotDiss = selectTOM^7
diag(plotDiss) = NA
TOMplot(plotDiss, selectTree, selectColors, main = "Network heatmap plot, selected
genes",col=gplots::colorpanel(250,'red',"orange",'lemonchiffon'))
##
MEs0 = moduleEigengenes(five, moduleColors)$eigengenes
# 计算根据模块特征向量基因计算模块相异度:
MEDiss = 1-cor(MEs0);
# Cluster module eigengenes
METree = hclust(as.dist(MEDiss), method = "average")
# Plot the result
plotEigengeneNetworks(MEs0,
"Eigengene adjacency heatmap",
marHeatmap = c(3,4,2,2),
plotDendrograms = FALSE,
xLabelsAngle = 90)

第七步:GO,KEGG富集分析

总结
- 因为一开始分的基因模块不一样,所以导致后面的结果也会出现差别。但最重要是过一遍WGCNA的流程,体会下作者如何描写图片的,这样就可以用在自己的文章中啦!
文末友情推荐
要想真正入门生物信息学建议务必购买全套书籍,一点一滴攻克计算机基础知识,书单在:什么,生信入门全套书籍仅需160 。
如果大家没有时间自行慢慢摸索着学习,可以考虑我们生信技能树官方举办的学习班:
- 数据挖掘学习班第7期(线上直播3周,马拉松式陪伴,带你入门),原价4800的数据挖掘全套课程, 疫情期间半价即可抢购。
- 生信爆款入门-第9期(线上直播4周,马拉松式陪伴,带你入门),原价9600的生信入门全套课程,疫情期间3.3折即可抢购。
如果你课题涉及到转录组,欢迎添加一对一客服:详见:你还在花三五万做一个单细胞转录组吗?
号外:生信技能树知识整理实习生招募,长期招募,也可以简单参与软件测评笔记撰写,开启你的分享人生!另外,:绝大部分生信技能树粉丝都没有机会加我微信,已经多次满了5000好友,所以我开通了一个微信好友,前100名添加我,仅需150元即可,3折优惠期机会不容错过哈。我的微信小号二维码在:0元,10小时教学视频直播《跟着百度李彦宏学习肿瘤基因组测序数据分析》