生存分析大家都会做了,单个或者多个基因表达量分组,肿瘤病人亚型分组策略,突变与否,多组学结合都可以分组后做生存分析。
看到2020年9月24日,俄罗斯喀山联邦大学的Igor Astsaturov发表文章Cholesterol Pathway Inhibition Induces TGF-β Signaling to Promote Basal Differentiation in Pancreatic Cancer,发现了胆固醇代谢调控胰腺导管癌发展和分化的机制,文章链接是:https://www.sciencedirect.com/science/article/pii/S1535610820304268
研究者们:we compared gene signatures between classical and basal subsets of PDAC using data from 76 high-purity (estimated >30% of transcripts originating from cancer cells) samples profiled by The Cancer Genome Atlas Research Network
针对挑选好的76个病人分组后,差异分析对基因进行排序,排序好的基因就可以跑GSEA,这里研究者选择的是:hallmark mRNA transcriptional signatures ,得到的结果如下:
差异分析,火山图,热图等等标准流程,基本上读一下我在生信技能树的表达芯片的公共数据库挖掘系列推文 就明白了:
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
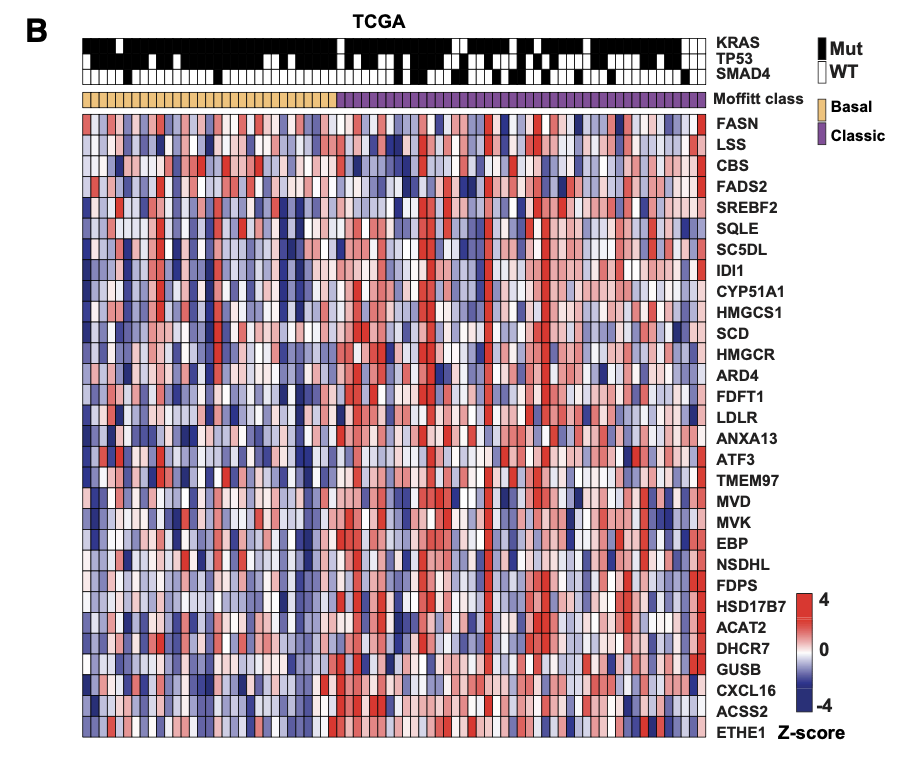
下面是一个多组学的可视化,加入了挑选好的76个病人的3个基因的突变信息,重点是展现 “hallmark cholesterol homeostasis” 这个基因集里面的基因。
(B) Heatmap of normalized expression of representative genes in the “hallmark cholesterol homeostasis” signature for TCGA cases.
可以看到,其实 “hallmark cholesterol homeostasis” 的全部基因并不是泾渭分明的在basal和classic两个组里面差异表达。
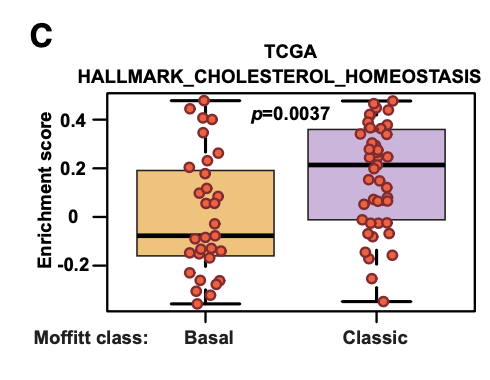
这个图C有点难以理解:(C) Comparison of “hallmark cholesterol homeostasis” gene signature in 76 PDAC cases from TCGA.
看起来应该是单样品的GSEA分析,就是ssGSEA啦。我在生信技能树多次讲解GSEA分析,见:- GSEA分析一文就够(单机版+R语言版)
- GSEA的统计学原理试讲
- GSVA或者GSEA各种算法都是可以自定义基因集的
- 基因集富集分析(GSEA)中的排序指标:它们重要吗?
- 200块的代码我的学徒免费送给你,GSVA和生存分析
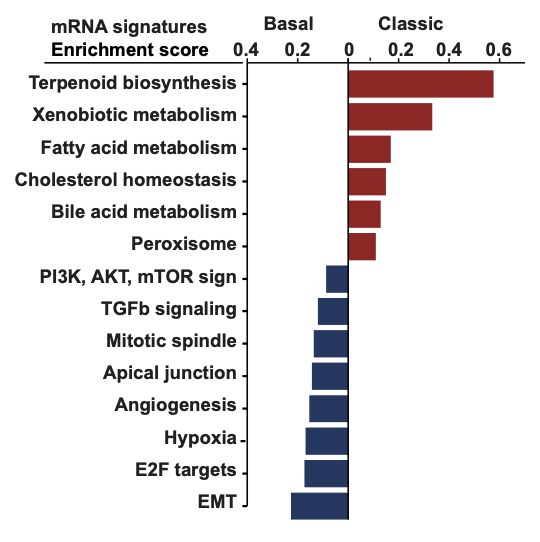
当然了,ssGSEA稍微有一点门槛:6. 纯R代码实现ssGSEA算法评估肿瘤免疫浸润程度
最后,神奇的事情发生了,根据这个“hallmark cholesterol homeostasis” signature打分,可以把76个病人首先分组,然后再继续分成basal和classic两个组,这个时候可以看到,肿瘤亚型在“hallmark cholesterol homeostasis” signature低的那个分组里面,生存分析是显著的!
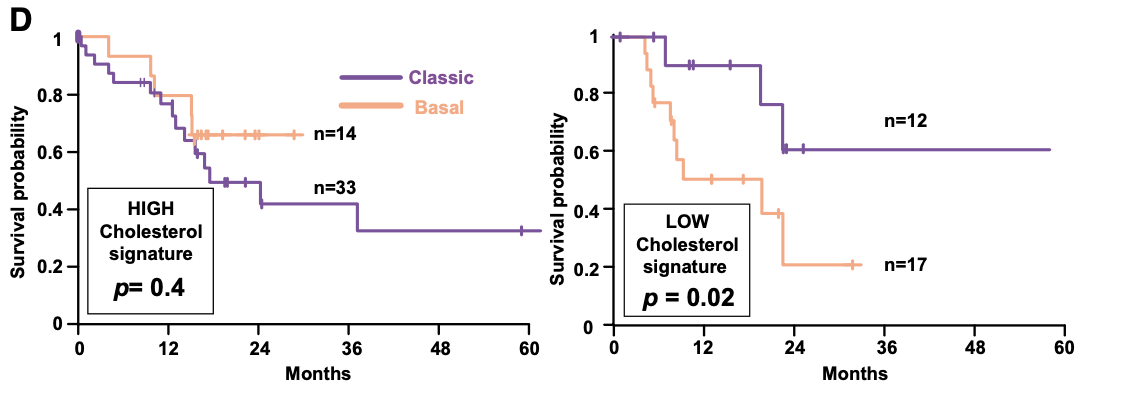
但是,问题就来了,可以看到前面的gsea分析里面,差异的基因集非常多哦,为什么作者定位到了“hallmark cholesterol homeostasis” signature呢?
莫非是作者批量跑了全部的其它通路,仅仅是“hallmark cholesterol homeostasis” signature具有隐藏分层的效应吗?
作为学徒作业
复现这个分析,首先在TCGA数据库里面找到76 high-purity (estimated >30%)的胰腺导管癌病人,然后根据basal和classic两个组进行差异分析,然后差异分析后的走gsea分析,以及单个样本的gsea分析。最后绘制出生存分析图表!