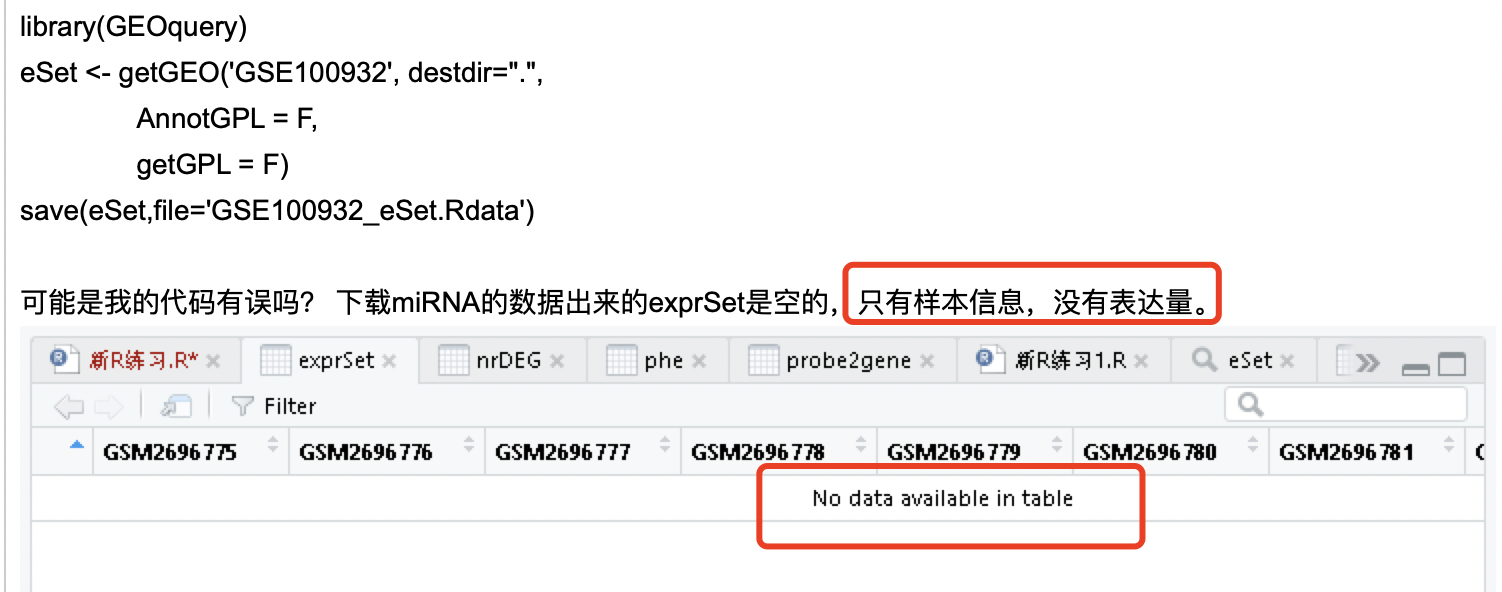
有粉丝咨询,他感兴趣的领域的一篇文章,提供了miRNA-seq数据,但根据我的GEO数据挖掘课程代码,没办法下载到表达量矩阵文件。如下:
希望我出一个miRNA-Seq数据实战视频课程,因为这篇文章的其原始数据上传到了公共数据库,见:https://www.ncbi.nlm.nih.gov/sra?term=SRP111349。恰好可以用来做课程示例。我首先指出来了粉丝的错误
使用我的GEO数据挖掘课程代码,我提供的例子是针对表达量芯片的,并不是针对测序数据。但并不是说,测序数据就不能分析,也并不是说它就没有提供表达矩阵。
文章对应的表达矩阵也是公开可以获取的,https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100932
其实这篇文章(URL: https://doi.org/10.1007/s12217-018-9653-2 )有3种数据:
- GSE100930 [microarray on total RNA]
- GSE100931 [RNA-seq]
- GSE100932 [miRNA-seq on miRNAs from exosomes]
文章的miRNA-seq数据分析
关于miRNA-seq数据分析流程流程,文献描述如下:
- Pre-processing: Sequencing adapters removal performed with Trimmomatic (v0.32)
- Pre-processing: reads in the size range of 18-50 nucleotides were selected and used for downstream analysis
- Mapping: mapped against mature miRNAs in the mirBase (version 21) using BWA tool (command bwa aln; v0.7.5a)
- Feature count: miRNAs expression counts were obtained with samtools (z1.2)
- Differential expression analysis: differential expression analysis was performed using the edgeR (Bioconductor version 2.13). Counts were normalized and differential expression was inferred using the negative binomial distribution and a shrinkage estimator for the distribution variance of the counts.
- Genome_build: mirBase: r21 mature RNA
- Supplementary_files_format_and_content: raw count of reads mapping to different miRNAs for each sample
多种多样的miRNA上游分析流程
欢迎共享哦,比如大家可以看到的tcga数据库的mRNA Analysis Pipeline ,详细代码:
- https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
太复杂的流程就算了,比如上面提到的发表在 Nucleic Acids Res. 2016 Jan 8; 的文章的流程:- https://github.com/bcgsc/mirna
普通人一辈子也就是处理两三次miRNA数据,并不是TCGA计划那样专业的团队,所以我们仅仅是关心测序reads的清洗问题,接头去除,以及比对的策略。定量之后的表达矩阵分析,反而是很简单的。欢迎分享你验证过的拿手的流程哈,发邮件给我,到 jmzeng1314@163.com文章的RNA-seq数据分析流程描述
前面我提到过,这个文章有3种数据,其中对应的RNA-seq数据分析流程描述如下:
- Pre processing: Sickle trimmer was used to remove low quality ends of the raw reads (using a quality threshold of 20, with option –q 20) and to remove short reads (using a minimum length threshold of 30, with option –l 30); Adapters were removed using CUTADAPT
- Mapping: Reads were firstly aligned to reference genome with TopHat2 (v2.1.1); unmapped reads were re-aligned with Bowtie2 (v2.2.9) in local mode. Alignments were joined with picard tools (v1.119)
- Feature count: Gene count data were obtained with htsseq-count command in HTSeq framework (v0.6.0)
- Differential expression analysis: performed using the edgeR (v3.16.5) package in R. After library normalization, the genewise exact tests for differences in the mean between the two groups of negative-binomially distributed counts was calculated with command exactTest. Significantly differentially expressed genes (DE-genes) between the two conditions (FMOM vs FMSM), without adjustment for multiple testing, were identified and exported for further analysis.
- Genome_build: ENSEMBLE GRCh38.p5 Human Genome assembly
- Supplementary_files_format_and_content: text file with mRNA count data.
下游分析千差万别
感兴趣可以细读表达芯片的公共数据库挖掘系列推文 ;
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
虽然表达矩阵的获取是类似的(不管是表达量芯片,还是测序), 但是下游分析千差万别:
而miRNA-seq数据虽然也是表达量矩阵,差异分析,差异miRNA列表,但是miRNA本身是没有后续分析的丰富的数据库资源的。之前我在生信技能树分享了几个miRNA的靶向基因的查询工具,分别是:- microRNAs靶基因数据库哪家强
- 使用miRNAtap数据源提取miRNA的预测靶基因结果
- 对miRNA进行go和kegg等功能数据库数据库注释
如果你把你的目标miRNA的靶基因找到了,就可以走差不多的下游分析,包括KEGG,GO等生物学功能数据库的注释。学徒作业
参考我B站的miRNA-Seq数据挖掘实战视频课程:
- 教学视频免费在B站:https://www.bilibili.com/video/BV1zK411n7qr
- 课程配套思维导图见:https://mubu.com/doc/7A3T8hpUlLv
- miRNA相关资料在:https://share.weiyun.com/k6ZbUg6H