前面我们提到了:表达矩阵是如何得到的(CNS图表复现10),有粉丝提问,既然都开始走RNA-seq数据的上游分析了,到Linux服务器操作了,难道仅仅是为了拿到表达矩阵文件吗?RNA-seq数据分析可以有很多啊,比如融合基因,可变剪切,甚至变异位点。
的确文章正文是注明了全部的肿瘤病人的所有的癌症测序样品的热点区别情况,就是图1的:(C) Circle plot of the clinically identified oncogenic driver (outer circle) and treatment time point (inner circle) for each sample.
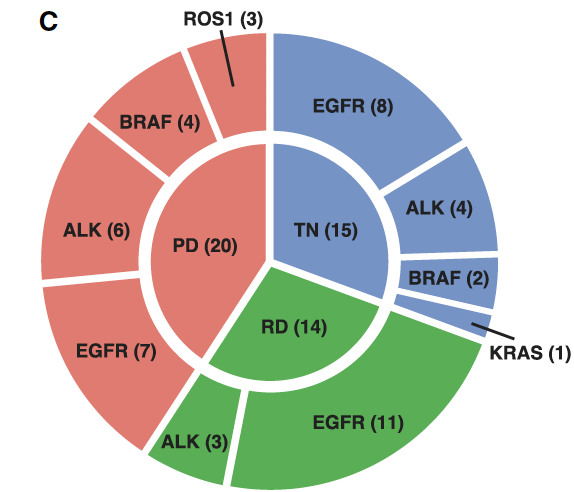
这个图的数据来源在文章的附件Excel表格,内容如下;
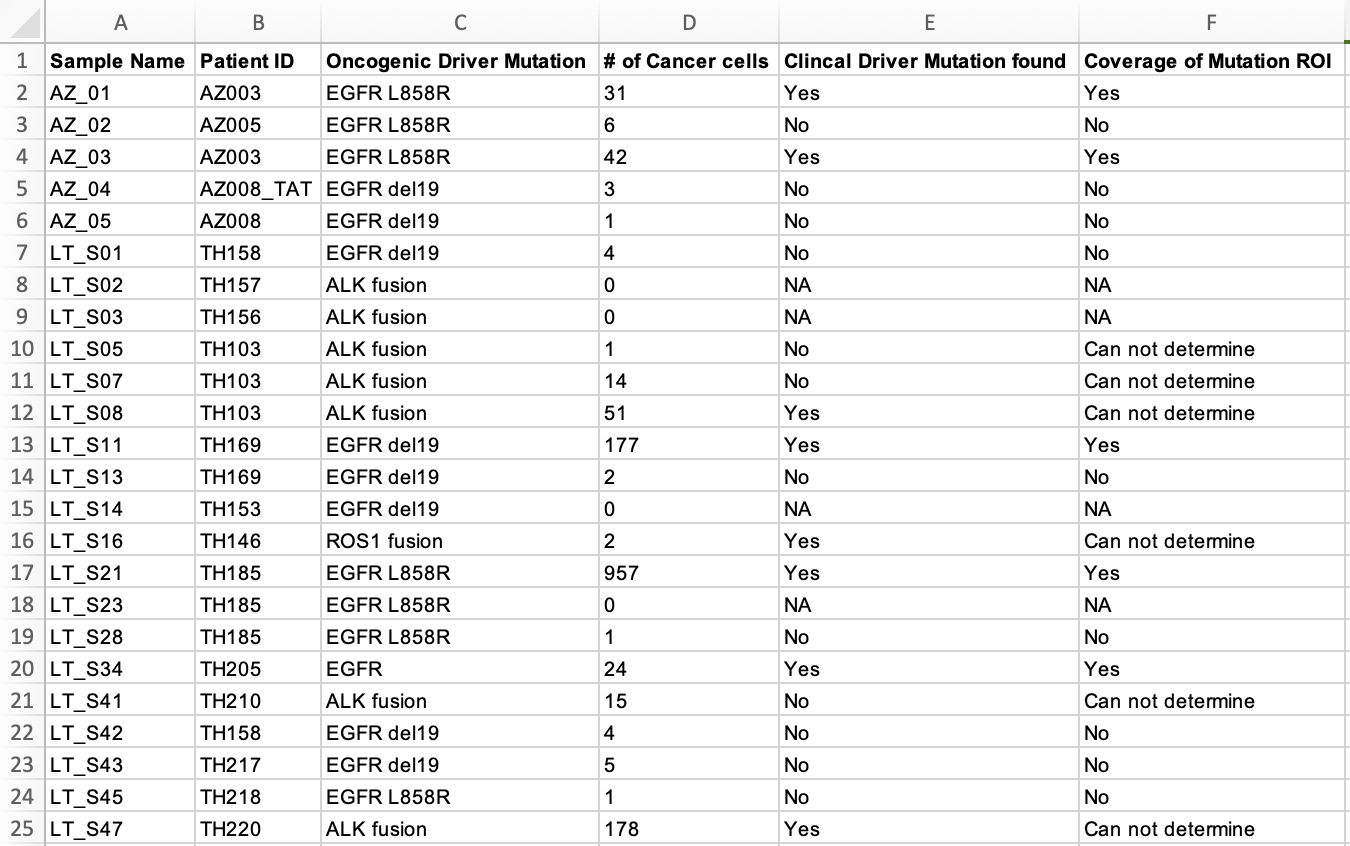
可以看到,每个病人的每个肿瘤样品是否含有突变位点,以及多少个癌症单细胞是含有这样的突变,都写的清清楚楚。
那么问题就来了,这个项目的单细胞RNA-seq数据肯定是可以推断具体的每个细胞是否有突变位点,不然就不会有上面的表格了!再具体看附件Excel表格,发现有更具体的信息,如下:
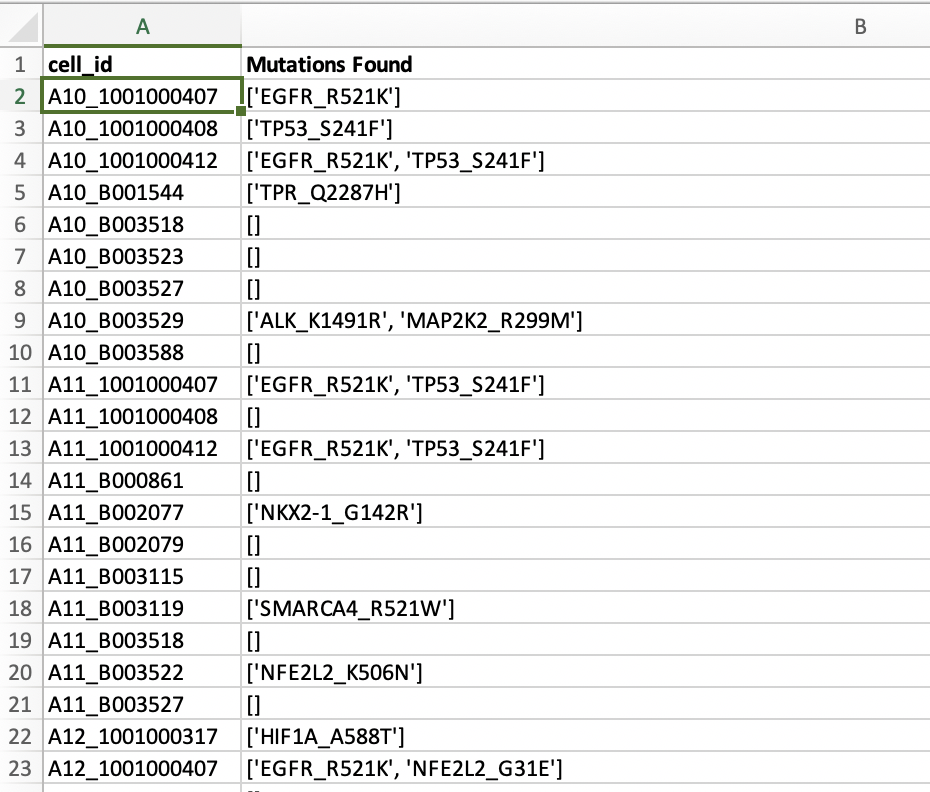
也就是说,每个细胞具体突变是什么,都列出一清二楚,而每个细胞我们只有单细胞RNA-seq数据,所以这个突变信息毫无疑问就是从这些单细胞RNA-seq数据拿到的!
RNA-seq数据找变异位点的最佳实践
很多人问针对RNA-seq数据如何找变异位点,尤其是肿瘤,比如TCGA计划的海量RNA-seq数据,所以我就写了教程:
- 2017年6月:RNA-seq 检测变异之 GATK 最佳实践流程
- 2019年11月:最新版针对RNA-seq数据的GATK找变异流程
并且分享了代码,就是STAR aligner 2-pass的比对,衔接上 GATK的MuTect2流程找变异位点。而且2018年6月发表在PeerJ的BIOINFORMATICS AND GENOMICS的文章标题是;《Detection and benchmarking of somatic mutations in cancer genomes using RNA-seq data》的文章,跟我的教程基本上差不多。
同样的,我们也是拿几个样品测试流程,样品的介绍在前面的教程:表达矩阵是如何得到的(CNS图表复现10)
conda activate rna
mkdir -p align
cd align
star_index=$HOME/biosoft/starFusion/db/GRCh38_gencode_v31_CTAT_lib_Oct012019.plug-n-play/ctat_genome_lib_build_dir/ref_genome.fa.star.idx/
ls -lh $star_index
bin_star=$HOME/biosoft/STAR-2.7.3a/bin/Linux_x86_64/STAR
## versionGenome 2.7.0d
gatk=$HOME/biosoft/gatk/gatk-4.1.8.1/gatk
DBSNP=$HOME/biosoft/GATK/resources/bundle/hg38/dbsnp_146.hg38.vcf.gz
kgSNP=$HOME/biosoft/GATK/resources/bundle/hg38/1000G_phase1.snps.high_confidence.hg38.vcf.gz
kgINDEL=$HOME/biosoft/GATK/resources/bundle/hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
bed=$HOME/annotation/CCDS/human/exon_probe.GRCh38.gene.150bp.bed
GENOME=$HOME/biosoft/starFusion/db/GRCh38_gencode_v31_CTAT_lib_Oct012019.plug-n-play/ctat_genome_lib_build_dir/ref_genome.fa
# $gatk CreateSequenceDictionary -R $GENOME # 最新版gatk,这个步骤非常快
for id in {15..19}
do
fq1=../clean/SRR107772${id}_1_val_1.fq.gz
fq2=../clean/SRR107772${id}_2_val_2.fq.gz
sample=SRR107772${id}
# star软件超级消耗内存,严格控制线程数量哦!
$bin_star --runThreadN 1 --genomeDir $star_index \
--twopassMode Basic --outReadsUnmapped None --chimSegmentMin 12 \
--alignIntronMax 100000 --chimSegmentReadGapMax parameter 3 \
--alignSJstitchMismatchNmax 5 -1 5 5 \
--readFilesCommand zcat --readFilesIn $fq1 $fq2 --outFileNamePrefix ${sample}_
mv ${sample}_Aligned.out.sam $sample.sam
samtools sort -o $sample.bam $sample.sam
samtools index $sample.bam
samtools flagstat $sample.bam > $sample.flagstat
# 这里需要判断上一个步骤是否成功,判断命令状态,简化脚本,就不写啦
# rm $sample.sam
sambamba markdup --overflow-list-size 600000 --tmpdir='./' -r $sample.bam ${sample}_rmd.bam
$gatk SplitNCigarReads -R $GENOME \
-I ${sample}_rmd.bam \
-O ${sample}_rmd_split.bam
# 因为我的star比对得到的bam文件里面没有 Read groups
# 参考:https://gatkforums.broadinstitute.org/gatk/discussion/6472/read-groups
$gatk AddOrReplaceReadGroups -I ${sample}_rmd_split.bam \
-O ${sample}_rmd_split_add.bam -LB $sample -PL ILLUMINA -PU $sample -SM $sample
$gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" BaseRecalibrator \
-I ${sample}_rmd_split_add.bam \
-R $GENOME \
-O ${sample}_recal.table --known-sites $kgSNP --known-sites $kgINDEL
$gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" ApplyBQSR \
-I ${sample}_rmd_split_add.bam -R $GENOME --output ${sample}_recal.bam -bqsr ${sample}_recal.table
bam=${sample}_recal.bam
$gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" HaplotypeCaller \
-ERC GVCF -L $bed -R $GENOME -I $bam --dbsnp $DBSNP -O ${sample}_gatk.gvcf
done
关于这个star软件安装,已经配套数据库文件的准备工作, 见:2019年11月:最新版针对RNA-seq数据的GATK找变异流程的教程。当然了,这个代码有点超纲了,对绝大部分初学者来说。需要把Linux的6个阶段跨越过去 ,一般来说,每个阶段都需要至少一天以上的学习:
- 第1阶段:把linux系统玩得跟Windows或者MacOS那样的桌面操作系统一样顺畅,主要目的就是去可视化,熟悉黑白命令行界面,可以仅仅以键盘交互模式完成常规文件夹及文件管理工作。
- 第2阶段:做到文本文件的表格化处理,类似于以键盘交互模式完成Excel表格的排序、计数、筛选、去冗余,查找,切割,替换,合并,补齐,熟练掌握awk,sed,grep这文本处理的三驾马车。
- 第3阶段:元字符,通配符及shell中的各种扩展,从此linux操作不再神秘!
- 第4阶段:高级目录管理:软硬链接,绝对路径和相对路径,环境变量。
- 第5阶段:任务提交及批处理,脚本编写解放你的双手。
- 第6阶段:软件安装及conda管理,让linux系统实用性放飞自我。
详见:《生信分析人员如何系统入门Linux(2019更新版)》
补充阅读;