在前面的教程 CNS图表复现05—免疫细胞亚群再分类 ,我提到到免疫细胞通常是以CD45阳性为标志,第一次分群规则是 :
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibo or CD31+,PECAM1,endo)
这样挑出来的immune (CD45+,PTPRC),细胞亚群继续细分可以成为: B细胞,T细胞,巨噬细胞,树突细胞等等。不过这个时候的分群就没有那么死板的规则了,很多灵活调整的余地。
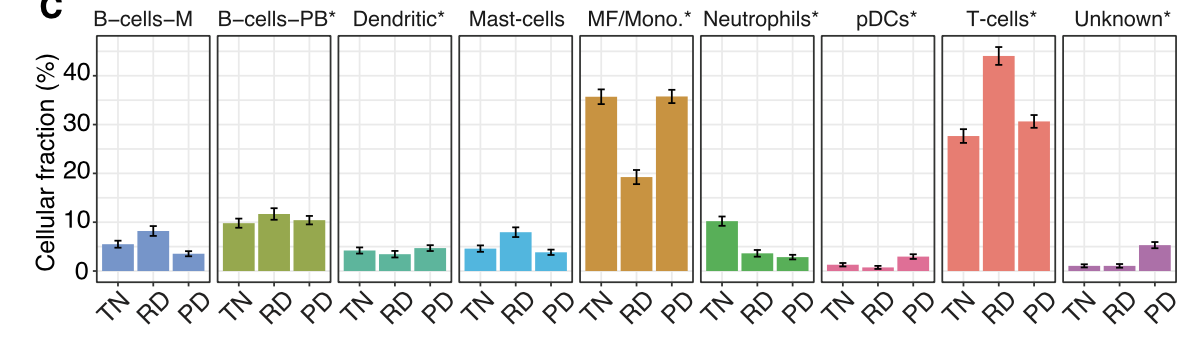
文章展示的一个简单的亚群分布如下:

作者依据自己的生物学背景做了一些自适应的调整, 见:CNS图表复现06—根据CellMarker网站进行人工校验免疫细胞亚群
我们也可以使用如下代码检查自己的免疫细胞细分亚群的结果:
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
library(ggplot2)
### 来源于:CNS图表复现02—Seurat标准流程之聚类分群的step1-create-sce.R
load(file = 'first_sce.Rdata')
### 来源于 step2-anno-first.R
load(file = 'phe-of-first-anno.Rdata')
sce=sce.first
table(phe$immune_annotation)
sce
cells.use <- row.names(sce@meta.data)[which(phe$immune_annotation=='immune')]
length(cells.use)
sce <-subset(sce, cells=cells.use)
sce
# 实际上这里需要重新对sce进行降维聚类分群,为了节省时间
# 我们直接载入前面的降维聚类分群结果,但是并没有载入tSNE哦
## 来源于: CNS图表复现05—免疫细胞亚群再分类
load(file = 'phe-of-subtypes-Immune-by-manual.Rdata')
sce@meta.data=phe
DimPlot(sce,reduction = "tsne",label=T, group.by = "immuSub")
table(phe$immuSub)
genes_to_check = c("PTPRC","CD19",'PECAM1','MME','CD3G', 'CD4', 'CD8A' )
p <- DotPlot(sce, features = genes_to_check,
assay='RNA',group.by = 'immuSub')
p
本来是23514个细胞,挑出来了13792个免疫细胞。然后,可以看到,单个基因都不能绝对的区分某个细胞亚群:

调整的空间还很大,一些标记基因混淆,其实亚群可以更加细致,然后合并同类亚群,总之实际操作会非常耗费时间,这里就不展开讲解了。并不需要与原文一模一样的。
查看各个单细胞的表型信息
这13792个免疫细胞的表型信息如下所示:
> as.data.frame(table(phe$biopsy_time_status))
Var1 Freq
1 Early 2550
2 off treatment 4068
3 PD 5389
4 PR 613
> as.data.frame(table(phe$biopsy_site))
Var1 Freq
1 Adrenal 566
2 Brain 9
3 Liver 1944
4 LN 3279
5 lung 1172
6 Lung 4850
7 Pleura 1972
通常情况下我们并不会比较不同免疫细胞的比例差异,就好像我们做基因表达量差异分析,很少会比较不同基因之间的表达量,因为基因本来就是不同的功能,管家基因的表达量本来就是超级高,无需跟其它基因比较。我们更关心的是同一个基因在不同的组的表达量变化,同理,我们想关心的也是不同的组的某个细胞亚群比例的变化。而在这个文章里面,就是:
首先我们可以使用前面的gplots包的balloonplot可视化方法:
library(gplots)
tab.1=table(phe$biopsy_time_status,phe$immuSub)
balloonplot(tab.1)
如下所示:
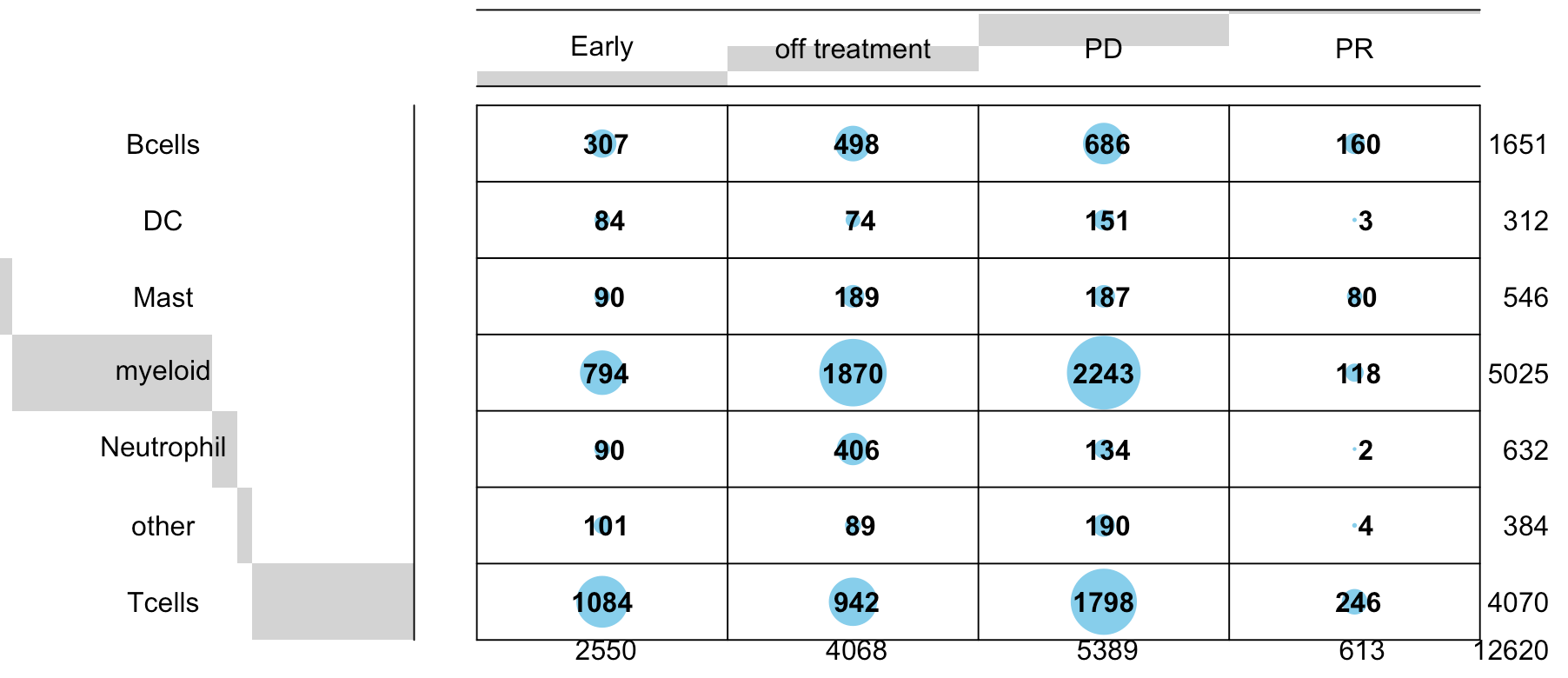
文章是 residual disease (RD) 和 on therapy progressive disease (PD),以及 patients before initiating systemic targeted therapy (TKI naive [TN]), 这3组。确实很诡异,但是作者提供的细胞表型列太多了了,看到我眼花缭乱,只好继续摸索,发现还有一个analysis列,就是3组:
ps=as.data.frame(table(phe$analysis,phe$biopsy_time_status))
ps=ps[ps$Freq>1,]
ps
> ps
Var1 Var2 Freq
1 grouped_pd Early 1129
2 grouped_pr Early 1421
6 naive off treatment 4068
7 grouped_pd PD 5389
11 grouped_pr PR 613
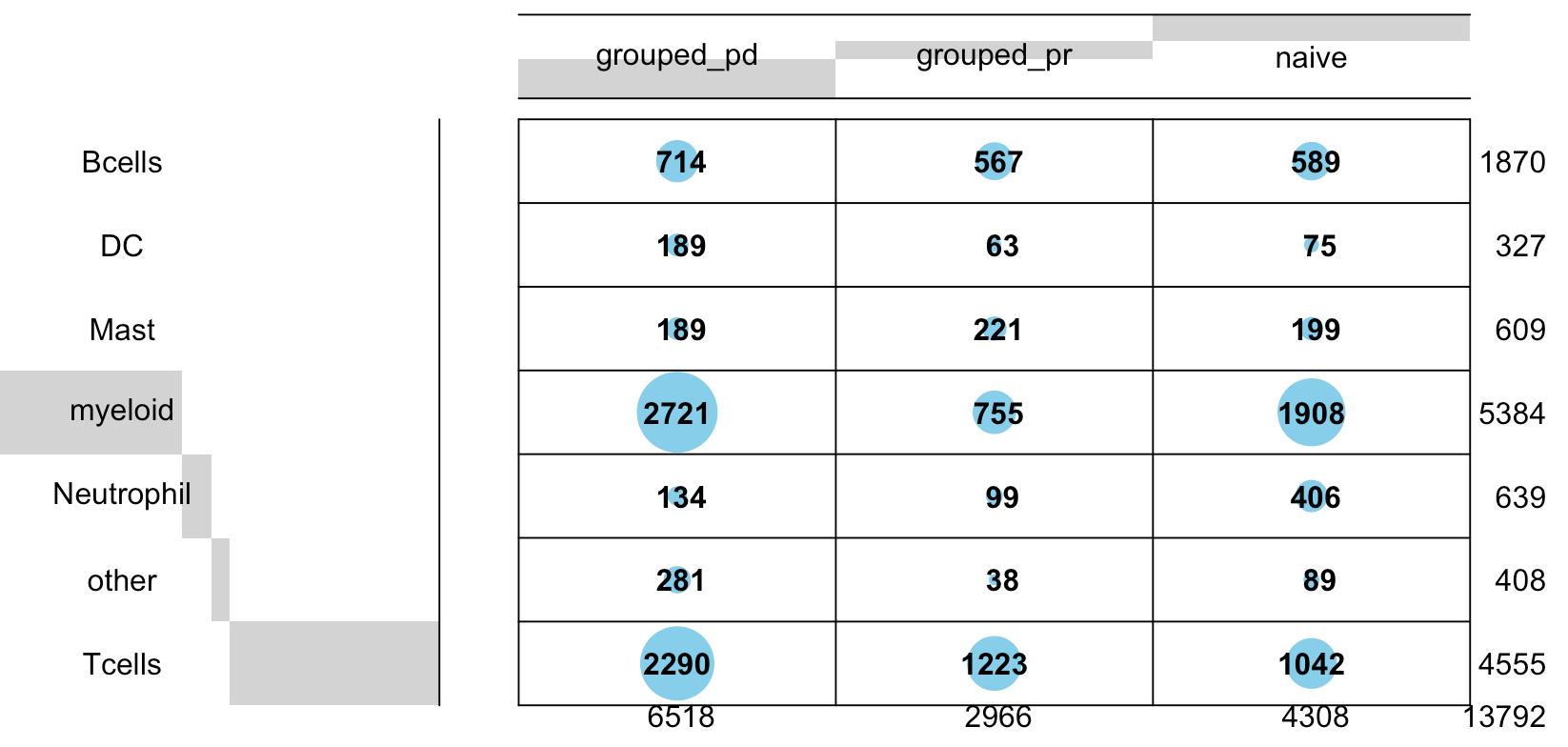
也就是说前面的Early组仍然是可以区分成为PD和PR组,再次出图如下:
原文给出来的是条形图,而且是比例,原图的代码是:
if(T){
library(ggrepel)
require(qdapTools)
require(REdaS)
#
metadata <- phe
# Keep only cells from tissues that are not brain or pleura
metadata <- metadata[-which(metadata$biopsy_site=="Brain" | metadata$biopsy_site=="Pleura"),]
# Convert to factor with logical order
metadata$analysis <- factor(metadata$analysis, levels = c("naive", "grouped_pr", "grouped_pd"))
# Create table and keep selected cell types
meta.temp <- metadata[,c("immuSub", "analysis")]
# Loop over treatment response categories
# Create list to store frequency tables
prop.table.error <- list()
for(i in 1:length(unique(meta.temp$analysis))){
vec.temp <- meta.temp[meta.temp$analysis==unique(meta.temp$analysis)[i],"immuSub"]
# Convert to counts and calculate 95% CI
# Store in list
table.temp <- freqCI(vec.temp, level = c(.95))
prop.table.error[[i]] <- print(table.temp, percent = TRUE, digits = 3)
#
}
# Name list
names(prop.table.error) <- unique(meta.temp$analysis)
# Convert to data frame
tab.1 <- as.data.frame.array(do.call(rbind, prop.table.error))
# Add analysis column
b <- c()
a <- c()
for(i in names(prop.table.error)){
a <- rep(i,nrow(prop.table.error[[1]]))
b <- c(b,a)
}
tab.1$analysis <- b
# Add common cell names
aa <- gsub(x = row.names(tab.1), ".1", "")
aa <- gsub(x = aa, ".2", "")
tab.1$cell <- aa
#
# Resort factor analysis
tab.1$analysis <- factor(tab.1$analysis, levels = c("naive", "grouped_pr", "grouped_pd"))
# Rename percentile columns
colnames(tab.1)[1] <- "lower"
colnames(tab.1)[3] <- "upper"
#
p<- ggplot(tab.1, aes(x=analysis, y=Estimate, group=cell)) +
geom_line(aes(color=cell))+
geom_point(aes(color=cell)) + facet_grid(cols = vars(cell)) +
theme(axis.text.x = element_text(angle = 45, hjust=1, vjust=0.5), legend.position="bottom") +
xlab("") +
geom_errorbar(aes(ymin=lower, ymax=upper), width=.2,position=position_dodge(0.05))
p1<- ggplot(tab.1, aes(x=analysis, y=Estimate, group=cell)) +
geom_bar(stat = "identity", aes(fill=cell)) + facet_grid(cols = vars(cell)) +
theme_bw() +
theme(axis.text.x = element_text(angle = 45, hjust=1, vjust=0.5), legend.position= "none") +
xlab("") +
geom_errorbar(aes(ymin=lower, ymax=upper), width=.2,position=position_dodge(0.05))
p1
}
首先,我觉得作者的数据转换代码很弱,其次绘图太复杂了,我就懒得解读了。
用我们的数据出图效果是:
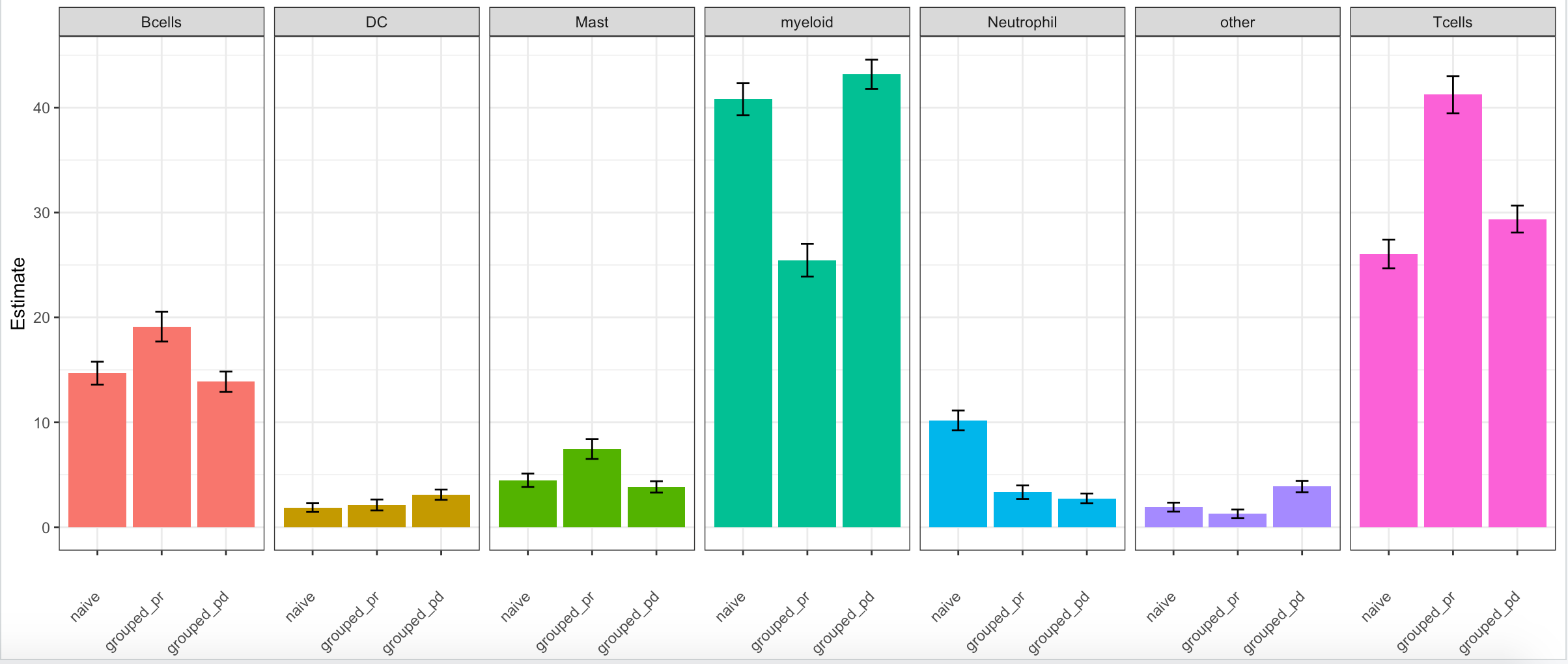
可以看到,其实与前面的gplots包的balloonplot可视化差不多效果。
也可以具体看病人:

主要是为了说明前面的细胞亚群比例变化,即使深入到具体的某个病人也是如此,绘图细节我这里 就不细讲了。
往期教程目录:
- CNS图表复现01—读入csv文件的表达矩阵构建Seurat对象
- CNS图表复现02—Seurat标准流程之聚类分群
- CNS图表复现03—单细胞区分免疫细胞和肿瘤细胞
- CNS图表复现04—单细胞聚类分群的resolution参数问题
- CNS图表复现05—免疫细胞亚群再分类
- CNS图表复现06—根据CellMarker网站进行人工校验免疫细胞亚群
- CNS图表复现07—原来这篇文章有两个单细胞表达矩阵
- CNS图表复现08—肿瘤单细胞数据第一次分群通用规则
- CNS图表复现09—上皮细胞可以区分为恶性与否
- CNS图表复现10—表达矩阵是如何得到的
- CNS图表复现11—RNA-seq数据可不只是表达量矩阵结果
- CNS图表复现12—检查原文的细胞亚群的标记基因
- CNS图表复现13—使用inferCNV来区分肿瘤细胞的恶性与否
- CNS图表复现14—检查文献的inferCNV流程
- CNS图表复现15—inferCNV流程输入数据差异大揭秘
- CNS图表复现16—inferCNV结果解读及利用
- CNS图表复现17—inferCNV结果解读及利用之进阶