前面我们公布了《cytof数据资源介绍(文末有交流群)》,现在就开始正式手把手教学。
上一讲我们提到了,跟着cytofWorkflow,可以使用read.flowSet函数全部的FCS文件后,会产生一个对象,这里面变量名是 samp 。其实cytofWorkflow只是一个流程而已,这个read.flowSet函数来自于R包。
但有了FCS文件不够,具体的每个样本是有临床表型的,而且呢,里面的抗体也是有对应的生物学意义的。这3种信息组合起来,就是 CATALYST 包的 prepData函数该派上用场啦,官方示例代码如下:
# construct SingleCellExperiment
library(CATALYST)
sce <- prepData(fs, panel, md, features = panel$fcs_colname)
官网链接是:
-
http://www.bioconductor.org/packages/release/bioc/vignettes/CATALYST/inst/doc/preprocessing.html
-
https://bioconductor.org/packages/release/bioc/vignettes/CATALYST/inst/doc/differential.html
读取抗体信息文件
首先我们可以看看cytofWorkflow的例子:
url <- "http://imlspenticton.uzh.ch/robinson_lab/cytofWorkflow"
# 这个cytof的panel的抗体信息表格:
panel <- "PBMC8_panel_v3.xlsx"
download.file(file.path(url, panel), destfile = panel, mode = "wb")
panel <- read_excel(panel)
head(data.frame(panel))
如下所示:
> head(data.frame(panel))
fcs_colname antigen marker_class
1 CD3(110:114)Dd CD3 type
2 CD45(In115)Dd CD45 type
3 pNFkB(Nd142)Dd pNFkB state
4 pp38(Nd144)Dd pp38 state
5 CD4(Nd145)Dd CD4 type
6 CD20(Sm147)Dd CD20 type
可以看到是24个抗体,每个抗体都有对应的名字以及分类。
构造临床表型变量
首先我们可以看看cytofWorkflow的例子:
library(readxl)
url <- "http://imlspenticton.uzh.ch/robinson_lab/cytofWorkflow"
md <- "PBMC8_metadata.xlsx"
download.file(file.path(url, md), destfile = md, mode = "wb")
md <- read_excel(md)
head(data.frame(md))
table(md[,3:4])
如下所示:
> head(data.frame(md))
file_name sample_id condition patient_id
1 PBMC8_30min_patient1_BCR-XL.fcs BCRXL1 BCRXL Patient1
2 PBMC8_30min_patient1_Reference.fcs Ref1 Ref Patient1
3 PBMC8_30min_patient2_BCR-XL.fcs BCRXL2 BCRXL Patient2
4 PBMC8_30min_patient2_Reference.fcs Ref2 Ref Patient2
5 PBMC8_30min_patient3_BCR-XL.fcs BCRXL3 BCRXL Patient3
6 PBMC8_30min_patient3_Reference.fcs Ref3 Ref Patient3
> table(md[,3:4])
patient_id
condition Patient1 Patient2 Patient3 Patient4 Patient5 Patient6 Patient7 Patient8
BCRXL 1 1 1 1 1 1 1 1
Ref 1 1 1 1 1 1 1 1
可以看到总共是16个FCS文件被读入了,来自于8个病人,每个病人都有两个不同的条件下的样品。
组合3个变量成为SingleCellExperiment对象
全部的代码如下:
require(cytofWorkflow)
library(readxl)
url <- "http://imlspenticton.uzh.ch/robinson_lab/cytofWorkflow"
md <- "PBMC8_metadata.xlsx"
download.file(file.path(url, md), destfile = md, mode = "wb")
md <- read_excel(md)
head(data.frame(md))
table(md[,3:4])
# 样本的表型信息
## 真正的表达矩阵
library(HDCytoData)
fs <- Bodenmiller_BCR_XL_flowSet()
# 如果网络不好,也可以自行下载
# 然后:loaded into R as a flowSet using read.flowSet() from the flowCore package
# 这个cytof的panel的抗体信息表格:
panel <- "PBMC8_panel_v3.xlsx"
download.file(file.path(url, panel), destfile = panel, mode = "wb")
panel <- read_excel(panel)
head(data.frame(panel))
# spot check that all panel columns are in the flowSet object
all(panel$fcs_colname %in% colnames(fs))
# 有了样本的表型信息,panel的抗体信息,以及表达量矩阵,就可以构建对象:
# specify levels for conditions & sample IDs to assure desired ordering
md$condition <- factor(md$condition, levels = c("Ref", "BCRXL"))
md$sample_id <- factor(md$sample_id,
levels = md$sample_id[order(md$condition)])
# construct SingleCellExperiment
library(CATALYST)
sce <- prepData(fs, panel, md, features = panel$fcs_colname)
就是使用 CATALYST 包的 prepData函数把3个信息整合起来哈。
如果你有自己的FCS文件,就需要使用read.flowSet函数读取,然后自己制作抗体表格,以及样本表型表格。必须要严格follow这里面的例子哦!
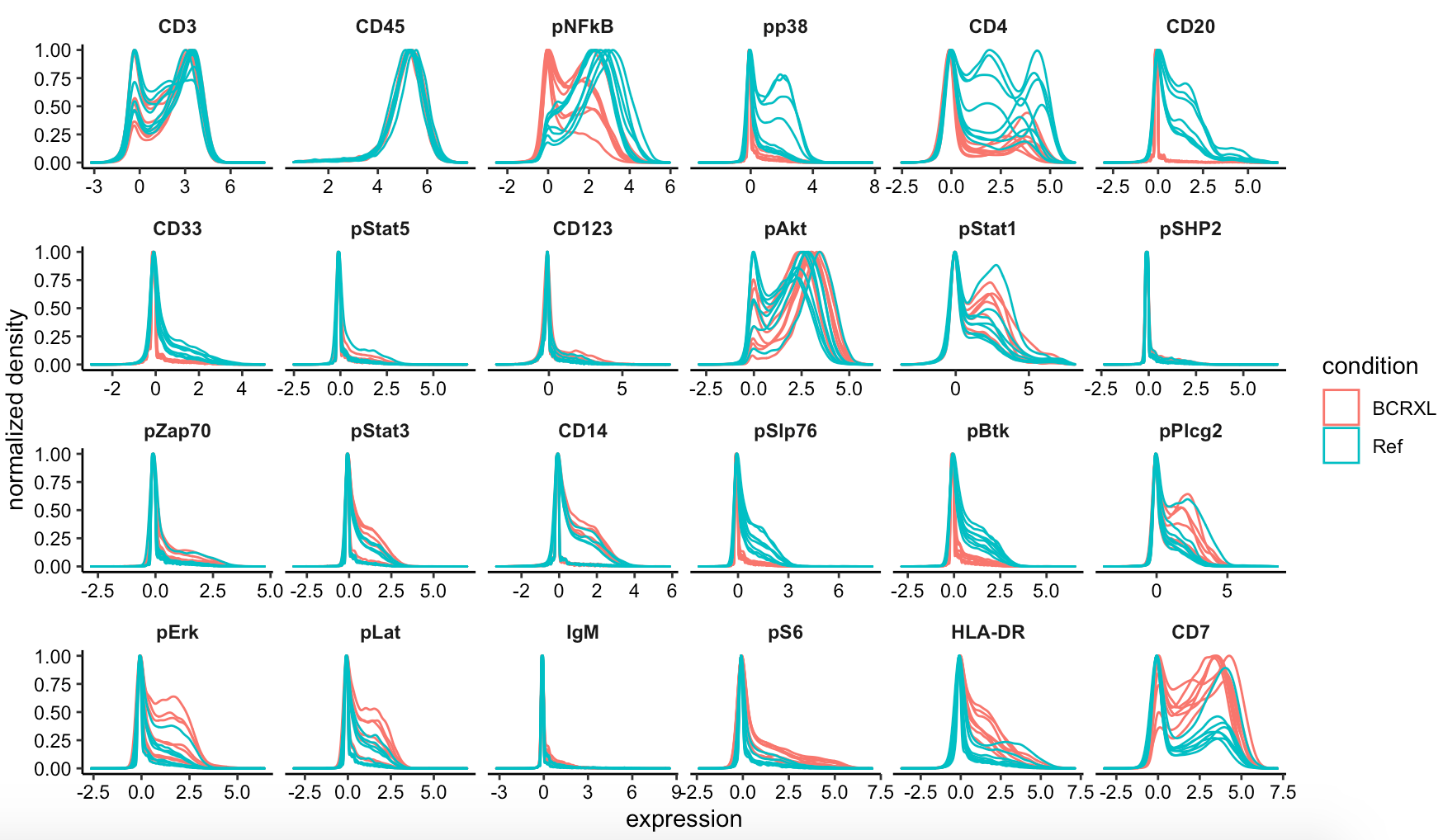
随便可以看看这些抗体在我们的不同病人的表达量分布情况:
require(cytofWorkflow)
p <- plotExprs(sce, color_by = "condition")
p$facet$params$ncol <- 6
p
密度图显示,信号值被归一化了: