最近读到发表在nature medicine杂志的文章《Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma》:
- 链接是:https://www.nature.com/articles/s41591-019-0694-x
它正文的 Data availability 部分就清晰地列出来了cytof数据和单细胞转录组数据存放的地方: - CyTOF data (Figs. 1, 2a,b, 3c–g and 4c,d) have been deposited with the FlowRepository (FR-FCM-Z2B3).
- scRNA-seq data (Figs. 2c–e and 3a) have been deposited with the Sequence Read Archive with accession number PRJNA588461.
在EBI可以下载到这个项目的10X单细胞转录组的测序数据
链接是:https://www.ebi.ac.uk/ena/browser/view/PRJNA588461 可以看到是7个样品的10X单细胞转录组的测序数据。每个样品是2个fq文件,所以可以毫无障碍的走我们的流程。参考我在《生信技能树》的教程:cellranger更新到4啦(全新使用教程)
目前单细胞转录组以10X公司为主流,我们也是在单细胞天地公众号详细介绍了cellranger流程,大家可以自行前往学习,如下:
- 单细胞实战(一)数据下载
- 单细胞实战(二) cell ranger使用前注意事项
- 单细胞实战(三) Cell Ranger使用初探
- 单细胞实战(四) Cell Ranger流程概览
- 单细胞实战(五) 理解cellranger count的结果
但是这个两年前的系列笔记是基于V2,V3版本的cellranger,目前呢它更新到了版本4,建议以我的最新版教程为准,在《生信技能树》的教程:cellranger更新到4啦(全新使用教程)
但是我在https://www.ncbi.nlm.nih.gov/sra?linkname=bioproject_sra_all&from_uid=588461看到了,它们的测序数据量介于 1~10G之间,远小于标准的100G数据量。
文章的数据分析描述如下: - These four separate count matrices were then merged into one large count matrix consisting of 13,263 cells (ranging from 2,763 to 3,666 cells per patient) by 19,187 genes.
- Next, the median number of unique molecules per cell was low across the four samples (1,170, 1,210, 1,468 and 1,592, respectively), resulting in a sparse data matrix, as is common to scRNA-seq data.
每个10X单细胞转录组的获得的细胞数量还算ok,基因数量也ok,但是呢,就是测序数据量有点偏低,很诡异!文章的单细胞转录组数据分析
单细胞转录组数据分析的细节,以及背景我就不赘述了,看我在《单细胞天地》的单细胞基础10讲:
- 01. 上游分析流程
- 02.课题多少个样品,测序数据量如何
- 03. 过滤不合格细胞和基因(数据质控很重要)
- 04. 过滤线粒体核糖体基因
- 05. 去除细胞效应和基因效应
- 06.单细胞转录组数据的降维聚类分群
- 07.单细胞转录组数据处理之细胞亚群注释
- 08.把拿到的亚群进行更细致的分群
- 09.单细胞转录组数据处理之细胞亚群比例比较
以及各式各样的个性化汇总教程,差不多就明白了。主要也是聚类分群
如下所示,针对的是TILs from untreated GBM tumors (n = 4),使用的是MAGIC算法进行聚类分群,并没有采用我们主推的seurat流程。但是仍然是以不同细胞亚群的标记基因热图来进行展示,如下:
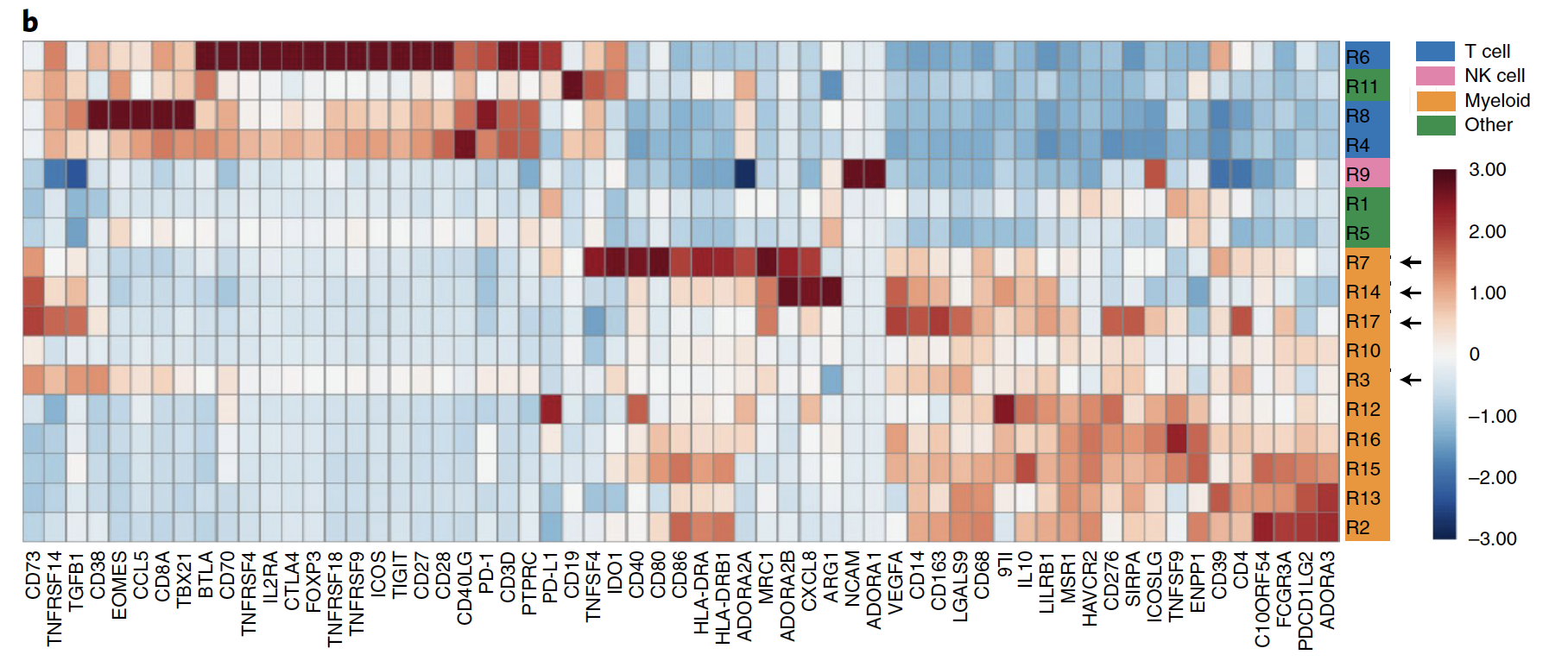
上图是:TILs from untreated GBM tumors (n = 4) were analyzed by scRNA-seq and identified using the MAGIC algorithm. Heatmap indicating the normalized expression of selected markers in leukocyte clusters identified by MAGIC.
但是,seurat实在是太全面了,仍然是推荐初学者从seurat开始认识单细胞数据分析的方方面面哈。我们《单细胞天地》的周运来大佬已经连夜翻译了他们的最新教程了,目录见: - Seurat 4.0 || 您的单细胞数据分析工具箱上新啦
- Seurat 4.0 || 单细胞多模态数据整合算法WNN
- Seurat 4.0 || 分析scRNA和膜蛋白数据
- Seurat 4.0 || WNN整合scRNA和scATAC数据
- Seurat 4.0 || 单细胞PBMC多模态参考数据集
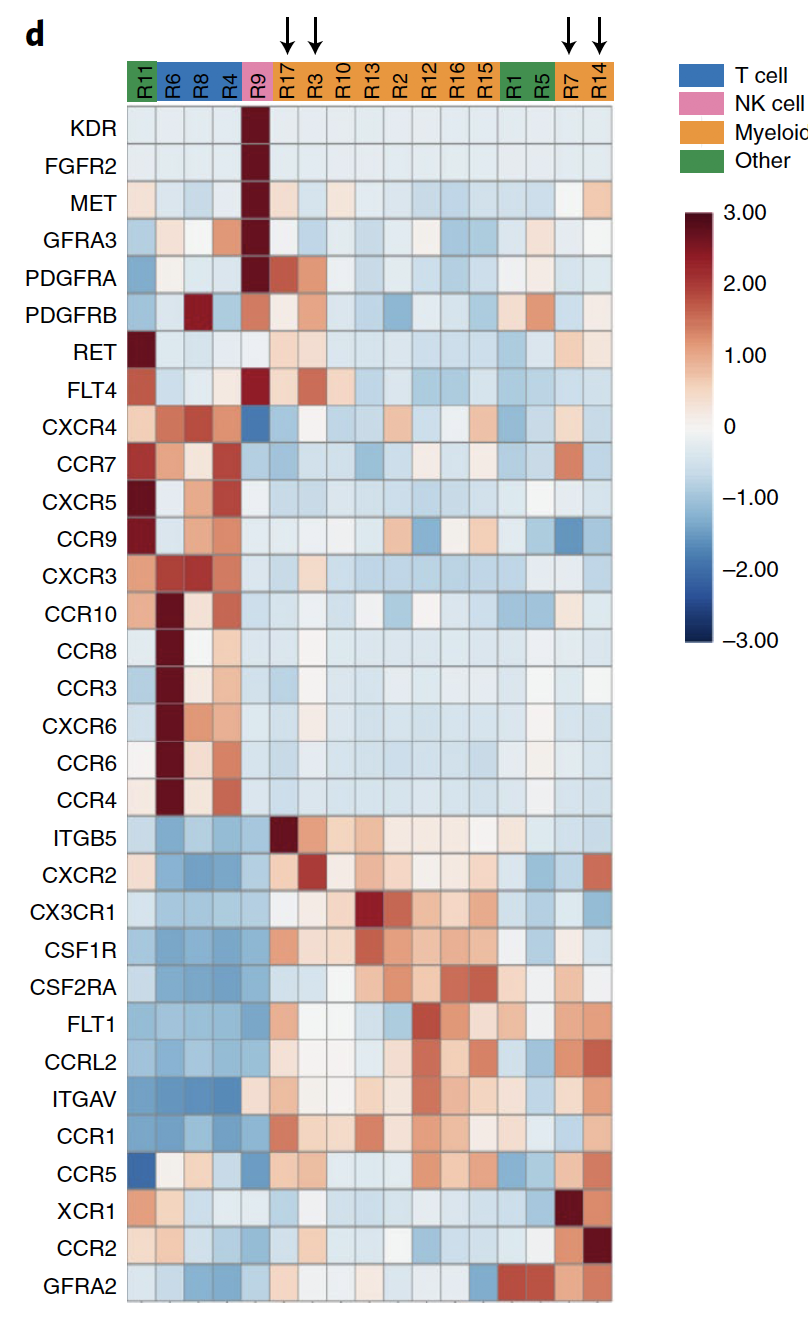
单独看 chemokine receptors基因集表达热图
前面的标记基因热图已经展示了不同的细胞亚群,接下来就是靠生物学背景了,比如,单独拿出来chemokine receptors基因集进行热图可视化,如下:
Heatmap indicating normalized expression of chemokine receptors on CD73hi macrophage clusters identified by MAGIC. 使用黑色箭头把 CD73hi myeloid 亚群标记了一下:
看基因表达量散点图
也是单细胞数据分析的常见图表之一,每个细胞首先被固定了坐标(tSNE或者umap的),然后可以在固定的坐标系对细胞属性进行不同的映射,表达量或者基因打分这样的连续性变量通常是映射为颜色深浅,点的大小。而细胞亚群这样的分类变量就映射为性状或者不同颜色。
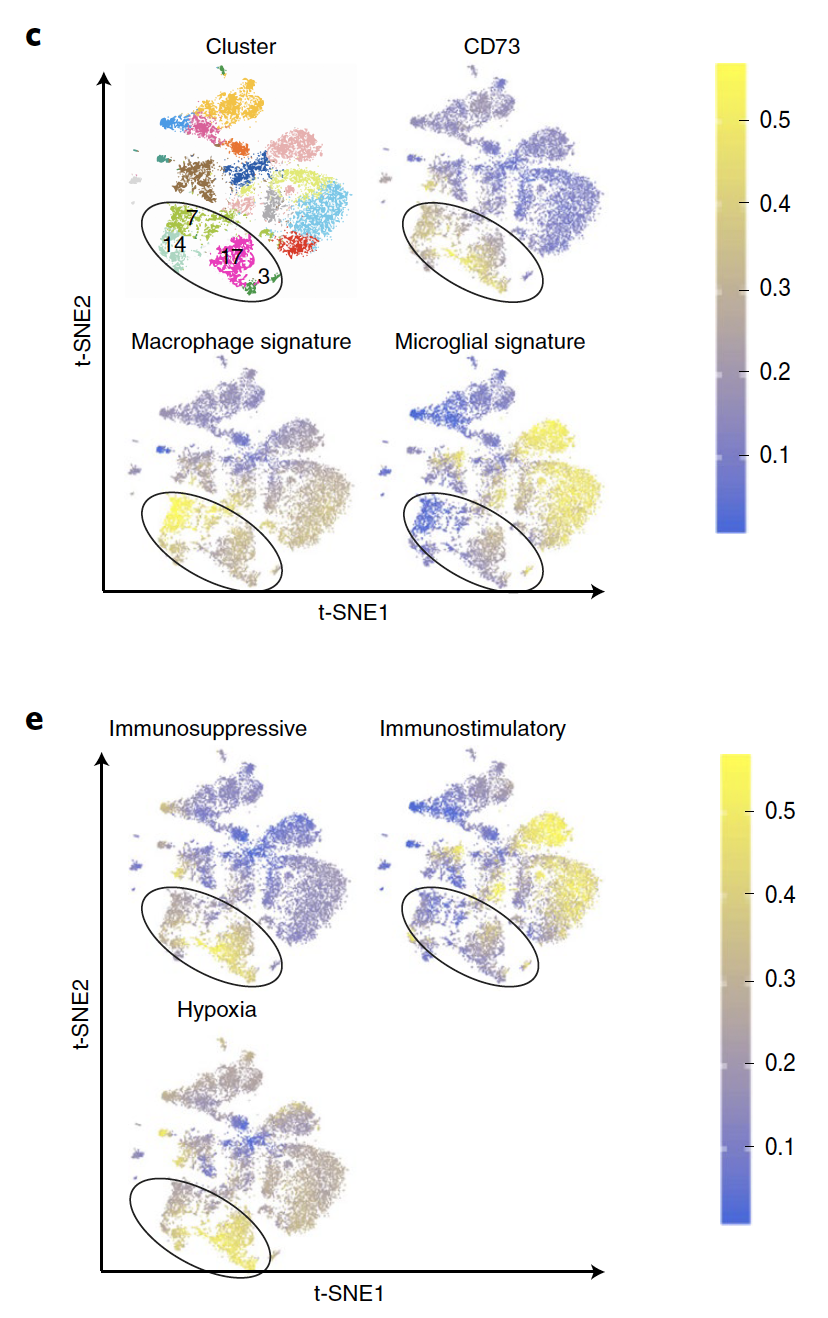
单独看macrophage gene signature 基因集表达热图
上面的基因表达量散点图,可以看到macrophage gene signature 基因集主要是在第 3,7,14,17亚群富集着,但是它展现的是macrophage gene signature 基因集做完一个整体的打分。既然作者这样的生物学结论都是关于macrophage gene signature 基因集的,就可以单独把其全部的基因拿出来后做热图进行更具体的可视化!
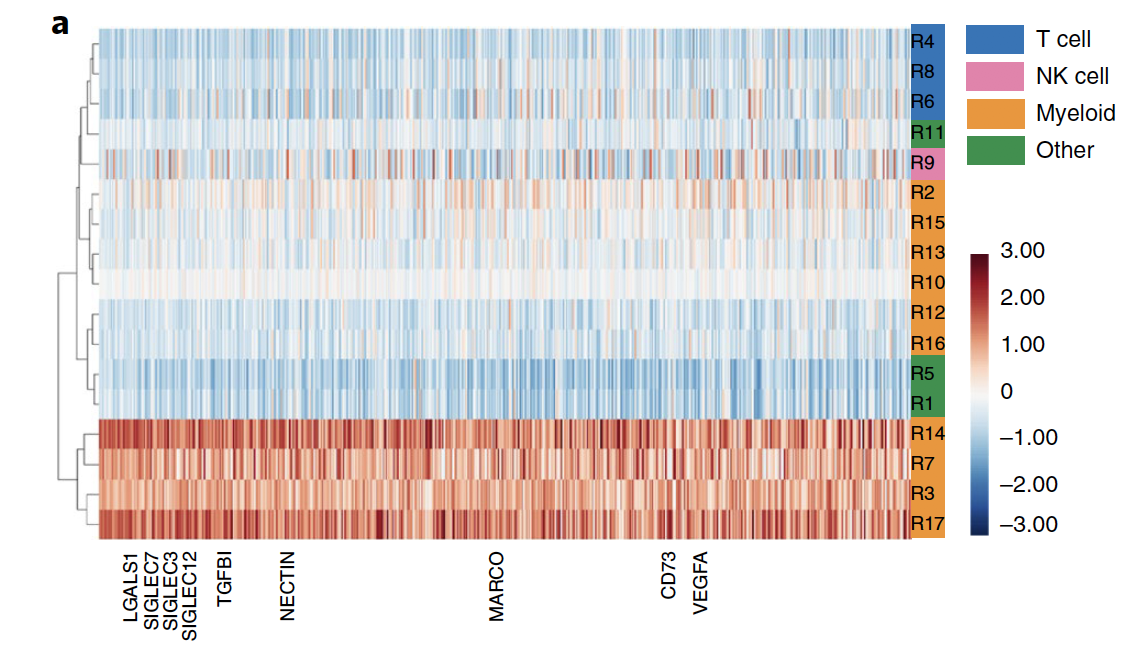
图例是: CD73hi macrophage gene signature of differentially expressed genes (z-score > 3.0, 45 genes; Supplementary Table 3). The heatmap indicates the normalized expression of top differentially expressed genes in CD73hi macrophages (z-score > 2.0) identified by MAGIC.学徒作业
当然是,根据文章的测序数据连接:https://www.ebi.ac.uk/ena/browser/view/PRJNA588461 ,下载7个样品的10X单细胞转录组的测序数据。每个样品是2个fq文件,走我在《生信技能树》的教程:cellranger更新到4啦(全新使用教程) ,每个样品拿到表达矩阵3个文件。
然后走下游seurat流程, 绘制上面的4个图!