众所周知,测序数据单端测序就是一个fq文件,双端测序就2个。但是呢,10X的单细胞转录组原始数据的话, 比较特殊,它的测序文库中包括index、barcode、UMI和测序reads。
- 首先,1-26个cycle就是测序得到了26个碱基,先是16个Barcode碱基,然后是10个UMI碱基;
- 然后,27-34这8个cycle得到了8个碱基,就是i7的sample index;
- 最后35-132个cycle得到了98个碱基,就是转录本reads,需要拿去做比对的!
然后呢,我们《生信技能树》目前出NGS数据处理教程,通常是会建议大家在EBI下载,这样的话,速度有保障!
比如文章Single-Cell RNA Sequencing of Lymph Node Stromal Cells Reveals Niche-Associated Heterogeneity. Immunity 2018 May 15;48(5):1014-1028.e6. PMID: 29752062 的表达矩阵在https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE112903 ,如果我们想从头到尾下载10X的单细胞转录组原始数据自己走cellranger流程,就需要注意数据库地址了。
在NCBI的SRA数据库如下,是两个样品的10X的单细胞转录组原始数据:

每个样品的10X的单细胞转录组原始数据都是3个文件,如下 :
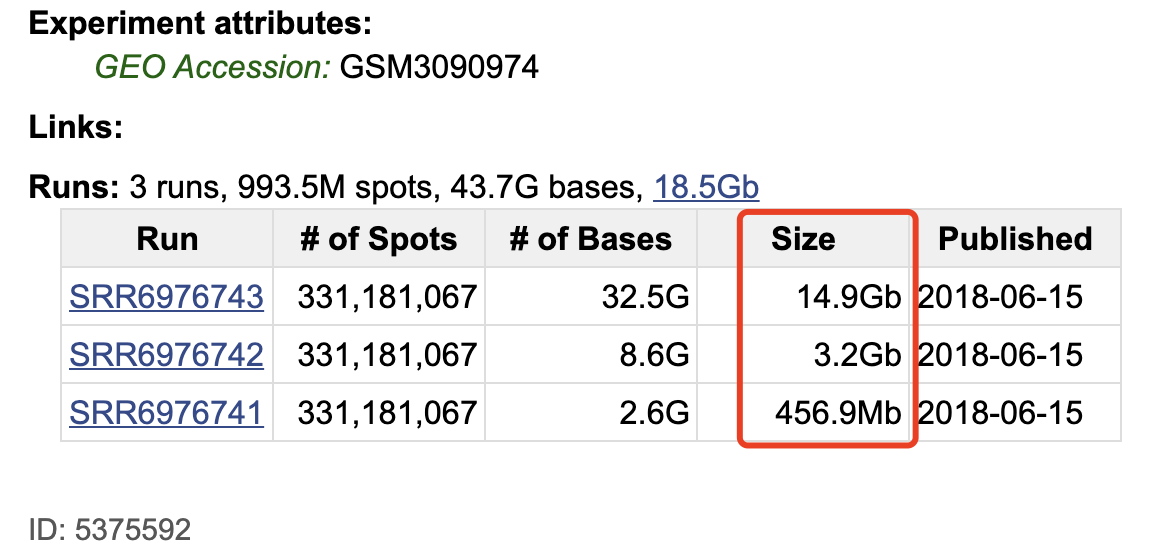
大家如果是从这个NCBI的SRA数据库下载,就会遇到速度超级慢的情况;
但是EBI就不一样
首先,ebi链接是:https://www.ebi.ac.uk/ena/browser/view/PRJNA449452 ,可以参考:使用ebi数据库直接下载fastq测序数据 , 需要自行配置好,然后去EBI里面搜索到的 fq.txt 路径文件。
很容易拿到全部的aspera链接:
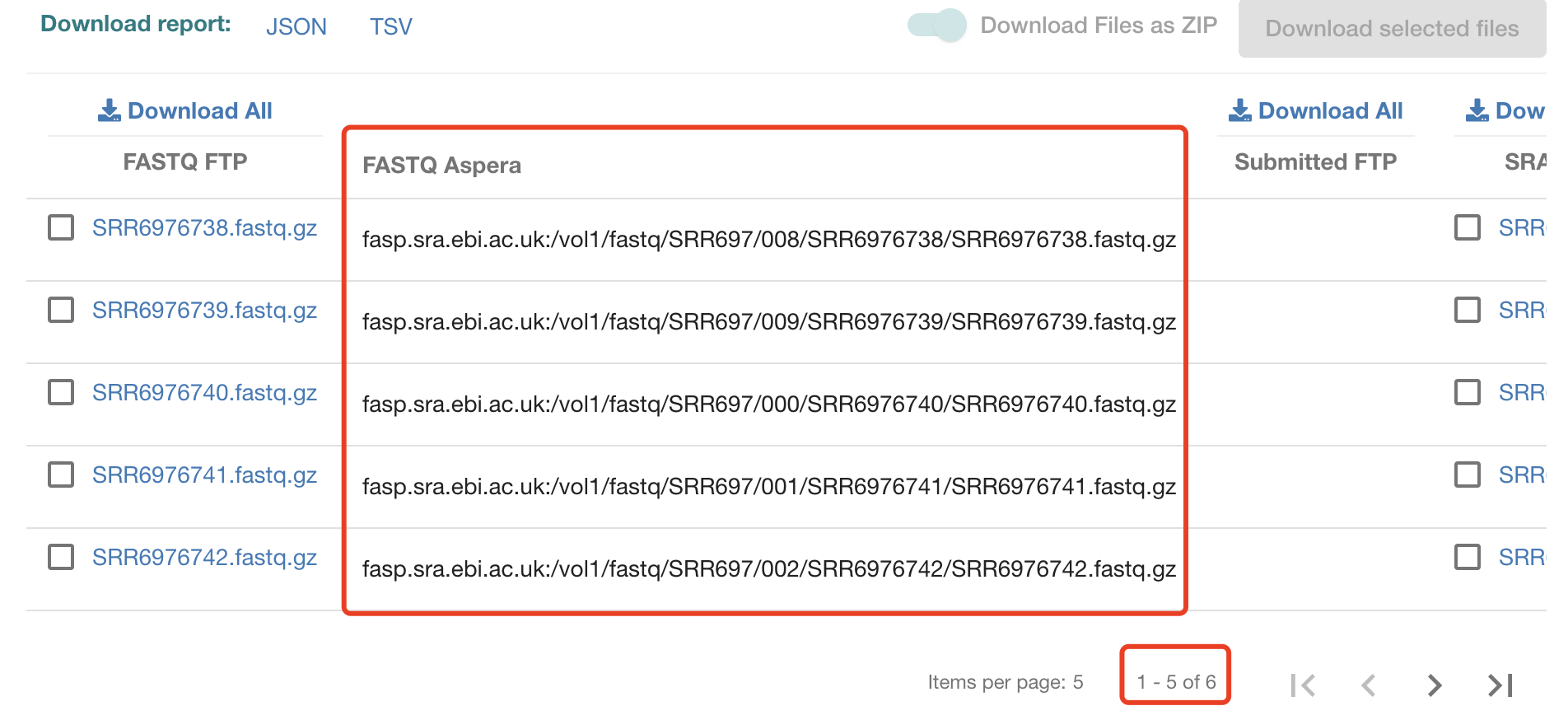
脚本如下:
# conda activate download
# 自己搭建好 download 这个 conda 的小环境哦。
cat fq.txt |while read id
do
ascp -QT -l 300m -P33001 \
-i ~/miniconda3/envs/download/etc/asperaweb_id_dsa.openssh \
era-fasp@$id .
done
# nohup bash step1-aspera.sh 1>step1-aspera.log 2>&1 &
这个脚本会根据你在EBI里面搜索到的 fq.txt 路径文件,来批量下载fastq测序数据文件。
这个fq.txt 路径文件内容如下:
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR697/008/SRR6976738/SRR6976738.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR697/009/SRR6976739/SRR6976739.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR697/000/SRR6976740/SRR6976740.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR697/001/SRR6976741/SRR6976741.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR697/002/SRR6976742/SRR6976742.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR697/003/SRR6976743/SRR6976743.fastq.gz
基本上一两个小时就下载了这100G的全部文件,如下所示:
2.2G 10月 31 19:59 SRR6976738.fastq.gz
6.1G 10月 31 19:59 SRR6976739.fastq.gz
21G 10月 31 19:59 SRR6976740.fastq.gz
2.2G 10月 31 19:59 SRR6976741.fastq.gz
6.1G 10月 31 19:59 SRR6976742.fastq.gz
21G 10月 31 19:59 SRR6976743.fastq.gz
全部下载成功后,需要 gzip -t 命令检查文件完整性。
然后需要文件重新命名哦!这个非常这样,官网给出的文件重新命名指南:https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/2.0/using/fastq-input#wrongname , 文件名不合格,程序肯定是失败的!
最后走我们的流程即可,参考我在《生信技能树》的教程:cellranger更新到4啦(全新使用教程)
目前单细胞转录组以10X公司为主流,我们也是在单细胞天地公众号详细介绍了cellranger流程,大家可以自行前往学习,如下:
- 单细胞实战(一)数据下载
- 单细胞实战(二) cell ranger使用前注意事项
- 单细胞实战(三) Cell Ranger使用初探
- 单细胞实战(四) Cell Ranger流程概览
- 单细胞实战(五) 理解cellranger count的结果
但是这个两年前的系列笔记是基于V2,V3版本的cellranger,目前呢它更新到了版本4,建议以我的最新版教程为准,在《生信技能树》的教程:cellranger更新到4啦(全新使用教程)
作为一个学徒作业吧
走完这个10X的单细胞转录组原始数据全部分析流程,拿到聚类分群图表,如下:
公共数据库下载的10X原始数据通常是有问题的
如果是一个SRA文件,解压成为3个文件:
2.6G 9月 19 13:21 SRR7722937.sra
593M 9月 19 13:21 SRR7722938_S1_L001_I1_001.fastq.gz
1.3G 9月 19 13:19 SRR7722938_S1_L001_R1_001.fastq.gz
3.4G 9月 19 13:22 SRR7722938_S1_L001_R2_001.fastq.gz
3.7G 9月 19 13:22 SRR7722938.sra
288M 9月 19 13:22 SRR7722939_S1_L001_I1_001.fastq.gz
606M 9月 19 13:22 SRR7722939_S1_L001_R1_001.fastq.gz
1.7G 9月 19 13:21 SRR7722939_S1_L001_R2_001.fastq.gz
1.8G 9月 19 13:22 SRR7722939.sra
329M 9月 19 13:22 SRR7722940_S1_L001_I1_001.fastq.gz
688M 9月 19 13:23 SRR7722940_S1_L001_R1_001.fastq.gz
1.7G 9月 19 13:20 SRR7722940_S1_L001_R2_001.fastq.gz
2.0G 9月 19 13:20 SRR7722940.sra
168M 9月 19 13:19 SRR7722941_S1_L001_I1_001.fastq.gz
350M 9月 19 13:20 SRR7722941_S1_L001_R1_001.fastq.gz
947M 9月 19 13:21 SRR7722941_S1_L001_R2_001.fastq.gz
1016M 9月 19 13:20 SRR7722941.sra
306M 9月 19 13:20 SRR7722942_S1_L001_I1_001.fastq.gz
646M 9月 19 13:20 SRR7722942_S1_L001_R1_001.fastq.gz
1.7G 9月 19 13:23 SRR7722942_S1_L001_R2_001.fastq.gz
这样的文件改名成功后就可以直接运行cellranger流程啦,通常是没有问题的。
但是,如果是EBI数据库下载3个文件:
2.2G 10月 31 19:59 SRR6976741.fastq.gz
6.1G 10月 31 19:59 SRR6976742.fastq.gz
21G 10月 31 19:59 SRR6976743.fastq.gz
它们的内容分别如下:
zcat SRR6976741_S1_L001_I1_001.fastq.gz |head
@SRR6976741.1 1/1
AGTGGAAC
+
AAA-<<JF
@SRR6976741.2 2/1
AGTGGAAC
+
AAAFFJJJ
以及
zcat SRR6976741_S1_L001_R1_001.fastq.gz |head
@SRR6976742.1 1/1
CTCGTCACAAATTGCCTGAACTGTCA
+
AAFF<JJJJJFJJJJJJFA-<FJFFJ
@SRR6976742.2 2/1
GTACGTACAATCACACTATTCCATCG
+
A-<<A<AAAAAF7<-FFJ<-F-<F<A
以及
zcat SRR6976741_S1_L001_R2_001.fastq.gz |head
@SRR6976743.1 1/2
AACNNNNCCNANACCCNTGGAGNNNGAGNCNANNTNNNNCNNANNNNTNTNGNTCNTGNGNGGATGAAGGTTGGGCTTAGTGAGGAGTGGGCGGCCGT
+
<-<####<-#A#FJ-F#--7F-###7A-#-#-##-####7##-####-#-#-#7-#7-#-#<-7---7---<A---7-<77A-<AF-----7------
@SRR6976743.2 2/2
GTCNNNNCGNANGCTGNTAGAANNNGTCNCNGNNTNNNNTNNTNNNNTNANGNTGNTANGNGTTATACGGTAGCTATCATTAAGAAGTAGGCGGAGGC
+
A<<####--#7#-A--#-7F--###7--#<#<##-####-##-####-#-#-#--#7A#-#7F77--7F--77---777-7<<-77-7----77-777
@SRR6976743.3 3/2
AATNNNNGANANANTANAATNNNNNNGTNTNTNNGNNNNTNNANNNNCNTNANTTNAGNANNCTGAACACACTATTTTGTATAAAGCCAATCTTAACG
可以看到,它们虽然是同一个样品的同一次测序,但是同一个reads被分割成为了3个文件后,reads的名字也被修改了。运行cellranger流程就会失败,需要把这3个文件内容也修改!
示例代码是:
mkdir new
nohup zcat SRR6976741_S1_L001_I1_001.fastq.gz | sed 's/SRR6976741./read/g'| \
gzip > new/SRR6976741_S1_L001_I1_001.fastq.gz &
nohup zcat SRR6976741_S1_L001_R1_001.fastq.gz | sed 's/SRR6976741./read/g'| \
gzip > new/SRR6976741_S1_L001_R1_001.fastq.gz &
nohup zcat SRR6976741_S1_L001_R2_001.fastq.gz | sed 's/SRR6976741./read/g'| \
gzip > new/SRR6976741_S1_L001_R2_001.fastq.gz &