常规的转录表达矩阵分析大家都应该是不陌生了,不管是芯片还是测序,最后都是表达矩阵走差异分析的流程。但这样仅仅是定位到基因集,生物学功能通路等等,如果讲一个完整的生物学故事有点单薄。以前我写过教程:基因集的转录因子富集分析,是一个算法层面的解决方案,不过在很多唯实验数据论的生物学家来看仍然是不够solid,其实可以加上一个真实的转录因子数据,我最近就看到了一个把转录组数据加上转录因子数据联合分析的非常好的文献。
发表在Cancer Cell 2019 Sep的文章 PMID: 31474569:《Single-Cell Transcriptomics in Medulloblastoma Reveals Tumor-Initiating Progenitors and Oncogenic Cascades during Tumorigenesis and Relapse. 》
关于Medulloblastoma (MB)
主要是有4个分子亚型:Wingless (WNT), Sonic hedgehog (SHH), Group 3, and Group 4
转录组数据的标准分析
具体分析可以看免费视频课程《RNA-seq数据分析》,这个研究者给出了如下所示的4张图:
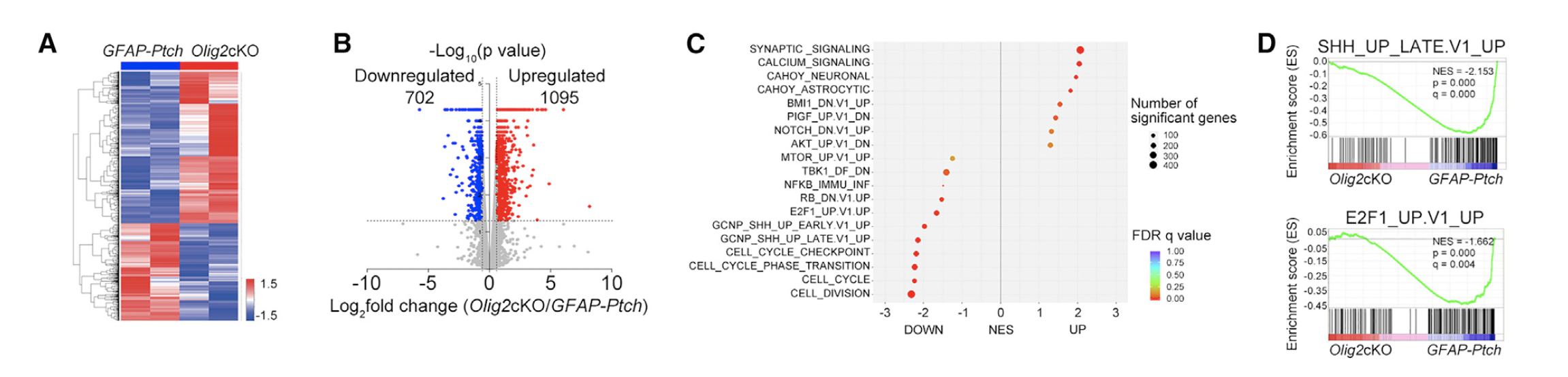
- (A) Heatmap of differentially expressed genes in Olig2cKO versus GFAP-Ptch tumors.
- (B) Volcano plot of significantly altered genes (p < 0.05).
- (C) Gene set enrichment analysis (GSEA) of top differentially regulated genes.
- (D) GSEA plots of downregulated SHH and E2F1 pathway gene sets in Olig2cKO versus GFAP-Ptch tumors.
数据在 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120974 ,如下的4个测序样品:
GSM3423064 RNA_GFAP_Ptch_1
GSM3423065 RNA_GFAP_Ptch_2
GSM3423066 RNA_GFAP_Ptch_Olig2_1
GSM3423067 RNA_GFAP_Ptch_Olig2_2
确实是非常标准的分析啦,差异分析后做出来火山图和热图,然后进行GSEA分析拿到一些生物学功能通路展示一下。但是很明显,研究者们并不满足于敲掉Olig2基因会导致SHH and E2F1 pathway失活这样的结论,仍然是想知道Olig2基因是如何调控这个过程的!
所以加上ChIP-seq数据
具体ChIP-seq数据分析可以免费视频课程《ChIP-seq数据分析》,研究者们给出来了如下所示4张图:
- (F) Heatmaps of OLIG2 and H3K27ac ChIP-seq signals in normal cerebellum (CB) and MB tumors. Box contains MB-specific target sites.
- (G) H3K27Ac ChIP-seq enrichment profiles around OLIG2-bound regions in MB tumors.
- (H) The most significantly enriched motifs in OLIG2-bound regions in MB tumors.
- (I) ToppGene analysis of OLIG2 and H3K27ac co-targeted pathways in MB tumors. hES, human embryonic stem cells.
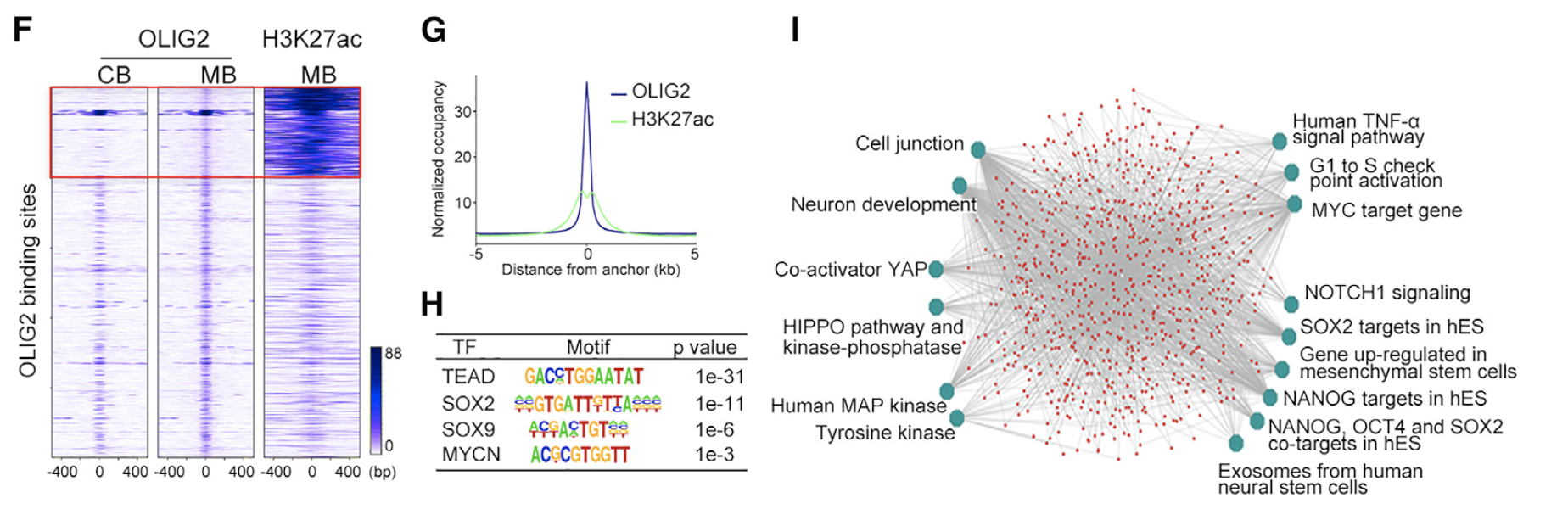
作者关心的就是OLIG2-bound regions in MB tumors。
数据在https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120968,9个样品:
GSM3423044 ChIP_CB_Olig2
GSM3423045 ChIP_MB_Olig2
GSM3423046 ChIP_MB_H3k27ac
GSM3936983 ChIP_CB_H3k27ac
GSM3936984 ChIP_MB_Olig2_repeat 2
GSM3936985 ChIP_MB_Olig2_repeat 3
GSM3936986 ChIP_MB_H3k27ac_repeat 2
GSM3936987 CB_Input
GSM3936988 MB_Input
可以看到CB和MB都是有各自独立的INPUT,这个是ChIP-seq实验设计的重中之重哦。
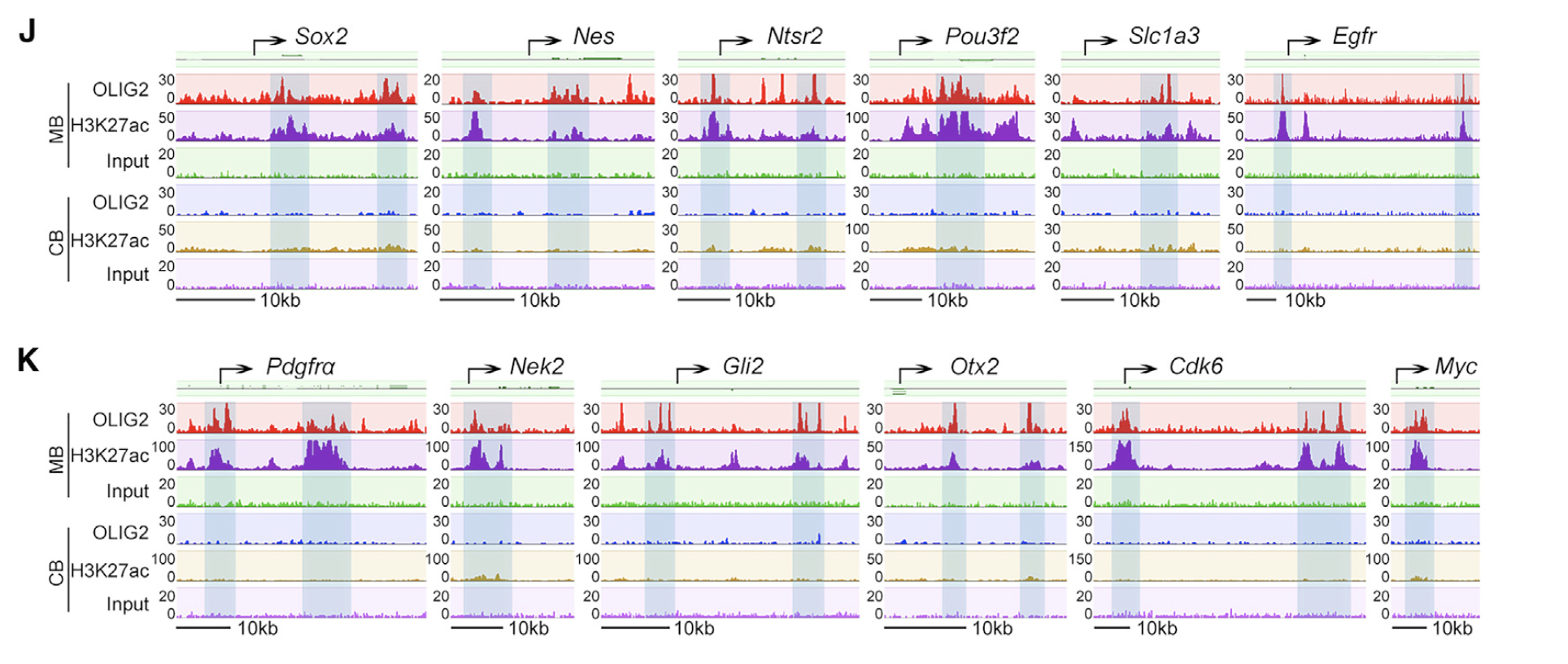
一些关键基因的IGV,包括:
- (J) OLIG2 and H3K27ac occupancy on genes associated with stemness.
- (K) OLIG2 and H3K27ac occupancy on genes associated with cell proliferation and on proto-oncogenes.
再加一个转录组呢
数据仍然是在 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120974 ,如下的6个测序样品:
GSM3936990 RNA_Olig2-GFP+_1
GSM3936991 RNA_Olig2-GFP+_2
GSM3936992 RNA_Olig2-GFP+_3
GSM3936993 RNA_Olig2-GFP-_1
GSM3936994 RNA_Olig2-GFP-_2
GSM3936995 RNA_Olig2-GFP-_3
这个时候作者并没有按常规通路分析,火山图,差异分析,富集分析,数据库注释等等,而是:
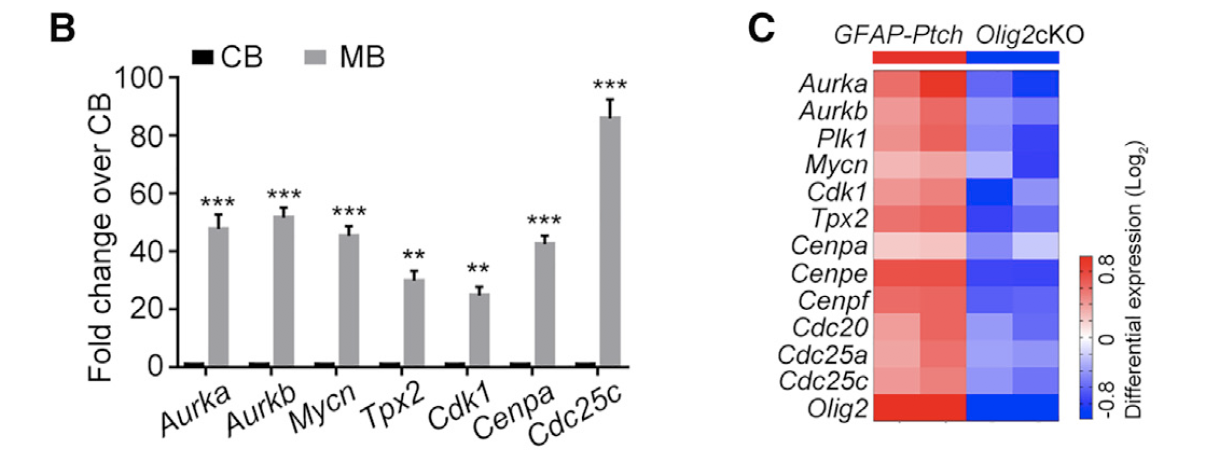
图例是:
- (B) qRT-PCR analyses of AURORA-A/MYCN pathway genes in GFAP-Ptch tumors.
- (C) Heatmap of expression of AURORA-A/MYCN pathway genes in Olig2cKO versus GFAP-Ptch tumors.
数据分析可以常规也可以个性化
这就是为什么绝大部分公司的流程化数据分析报告对大家的课题帮助并不是很大,因为并不是所有的的数据都会使用常规分析结果。一定会结合大家各自的生物学背景,进行各式各样的定制化分析。最后,还是推荐大家都学一点数据分析,B站免费NGS数据处理视频课程值得你学习,已经组建了微信交流群的有下面这些: