翻译自:https://constantamateur.github.io/2020-10-24-scBatch2/
更多教程见其博客: https://constantamateur.github.io/2020-04-10-scDE/
为什么 bulk RNA-seq 差异表达在单细胞世界中不是最有用的
2020/4/10
试图找出哪些基因在不同条件下“不同”,这可能是任何人想要进行 bulk RNA-seq 实验的第一件事。
当我们说“不同”时,实际上是指哪些基因在表达上的差异大于随机统计波动所预期的差异。这种变化的大小通常表示为条件之间标准化表达值的对数倍变化,并且统计波动采用负二项式分布进行参数化。用于处理 bulk RNA-seq(例如 edgeR 或 DESeq2 )的工具非常强大且成熟。
因此,当单细胞 RNA-seq 出现时,能将相同的想法应用于新环境。当然,也有许多尝试使用不同的统计分布和假设来更好地适应单细胞数据的属性。快速浏览一下谷歌就会发现,有足够多的相互竞争的方法可以保证至少有一篇基准论文。
作者认为匆忙为单细胞“更好”做差异表达而忽略的一件事是,被问到的生物学问题有细微的差别。或更确切地说,我们作为科学家最关心的结果并不是那些为批量数据开发的工具所激发的传统方法所强调的结果。
bulk RNA-seq 实验中差异表达的基因代表条件之间大细胞聚集体中总表达水平的变化。因此,条件之间的表达倍增是重要且有意义的,因为它告诉我,在一种条件下表达基因 A 的细胞大约是另一种条件下表达基因 A 的细胞的两倍。对于单细胞“差异表达”,大多数时候我真正想知道的是哪些基因在一组细胞中表达,而在其他任何地方都没有。
假设我正在将肿瘤细胞与周围的正常组织进行比较。一个基因 A 在每个肿瘤细胞中大约有 3 个拷贝,在周围的内皮细胞中大约有 1 个拷贝,这有点含糊而有趣。但是您不能针对一种药物,也不能通过免疫组化或 smFISH 进行很好的验证为“高表达”。真正想要找到的是像 CA9 这样的基因,它们在肾癌细胞中表达,而不在其他细胞中表达。
为了说明这一点,作者下载了 10X PBMC数据 具有包含注释和 tSNE 坐标 SoupX。
library(Matrix)
#Load PBMC data and make barcodes consistent with SoupX annotation
dat = readMM('filtered_gene_bc_matrices/GRCh38/matrix.mtx')
rownames(dat) = read.table('filtered_gene_bc_matrices/GRCh38/genes.tsv',sep='\t',header=FALSE)$V2
colnames(dat) = read.table('filtered_gene_bc_matrices/GRCh38/barcodes.tsv',sep='\t',header=FALSE)$V1
colnames(dat) = gsub('-1','',colnames(dat))
#Load tSNE co-ordinates and clustering recorded in SoupX
library(SoupX)
data(PBMC_DR)
B细胞是簇 4 和 7 。使用 edgeR 查找与其余细胞相比 DE 的基因。这基本上只是快速入门 edgeR 分析,但是我们prior.df=0 之所以设置是因为我们有大量信息来计算过度分散,而无需在基因之间共享信息。
library(edgeR)
dge = DGEList(as.matrix(dat),group=ifelse(PBMC_DR$Cluster %in% c(4,7),'B','Other'))
mm = model.matrix(~group,data=dge$samples)
dge = estimateDisp(dge,mm,prior.df=0)
fit = glmQLFit(dge,mm)
fit = glmQLFTest(fit,coef=2)
tt = topTags(fit,n=20)
print(tt)
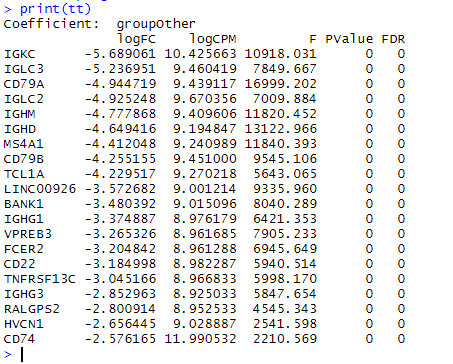
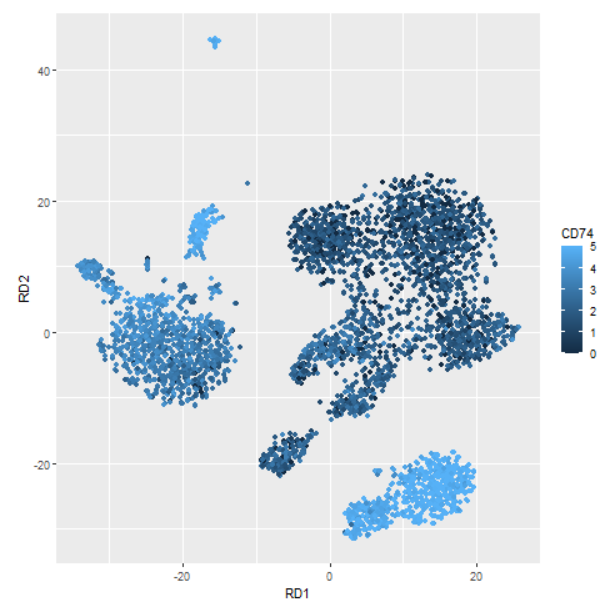
这些基因大多数被认为对B细胞非常特异,可以作为良好的标记物。可能应该选择一种不太明显的细胞类型来说明这一点。一个确实能说明作者观点的基因是 CD74 。如果我们绘制 CD74 的表达,我们会看到 CD74 在B细胞中最高(右下图),但它通常无处不在。这些是真正差异表达的基因,不一定很有趣。
normDat = log(1+1e4*t(t(dat)/colSums(dat)))
dd = PBMC_DR
dd$CD74 = normDat['CD74',]
library(ggplot2)
ggplot(dd,aes(RD1,RD2)) +
geom_point(aes(colour=ifelse(CD74>5,5,CD74))) +
labs(colour='CD74')
这只是挑剔吗? edgeR 发现的顶级基因列表确实非常好,如果您感兴趣的话,可以很容易地找到并过滤出上述无效的基因。
quickMarkers 在 SoupX 包的函数实现 tf-idf 方法
作者写这篇文章的动机不是挑剔 tools 例如 edgeR ,但要指出的是,如果您实际上只是对簇中最具体的基因感兴趣,则其他方法可能更合适。作者已经做了一段时间的比较怪异的事情是使用 tf-idf 的自然语言处理概念来获取特定于每个簇的基因的排序列表。这对于注释和理解许多单细胞数据集(例如本肾脏论文)非常有效,并且具有一些优势。
tf-idf 背后的想法是,它可以用于识别文档集合(称为语料库)中哪个单词最特定于一个文档。转化为转录组学,这等同于鉴定相对于所有其他细胞而言,哪些基因最特定于细胞簇。这有一些限制;如果要得到这样的结果,我们必须将 scRNA-seq 设为二进制。但是,它捕捉了我们在单细胞数据上进行“差异表达”时最经常感兴趣的本质。这种 tf-idf 方法是 quickMarkers 在 SoupX 包的函数中实现的。在 B 细胞上运行很简单,
mrks = quickMarkers(dat,ifelse(PBMC_DR$Cluster %in% c(4,7),'B','Other'),N=20)
mrks[mrks$cluster=='B',]
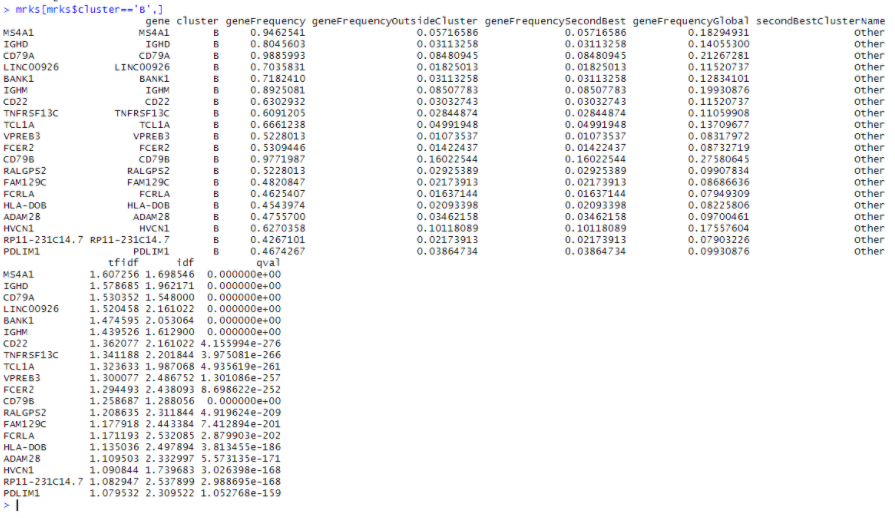
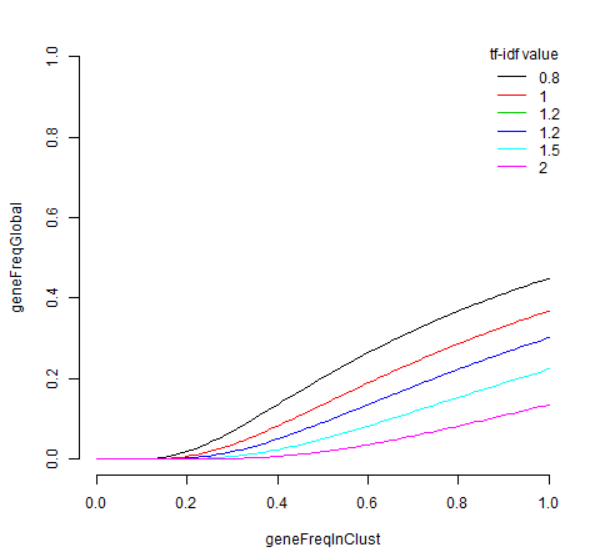
首先,此列表与 edgeR 列表之间存在很多重叠,前20个中有14个重叠。CD74 不在 quickMarkers 列表中,quickMarkers 列表中排名靠前的基因是“ MS4A1”(又名 CD20 ),这几乎是存在的最典型的 B细胞基因 。我发现额外的元数据列也非常有用。它们通常会告诉你每个基因在“兴趣簇中的哪部分细胞表达”和“兴趣簇之外的细胞表达”的空间中的位置。有趣的是,Tf-IDF 值在这个空间中具有这样的性质,即 Tf-IDF 值为 t 的基因满足
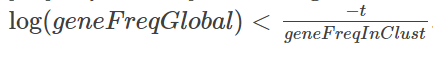
我们可以为此画几条曲线,
x=seq(0,1,.01)
plot(0,
type='n',
xlim=c(0,1),
ylim=c(0,1),
frame.plot=FALSE,
xlab='geneFreqInClust',
ylab='geneFreqGlobal')
ts = c(0.8,1,1.2,1.2,1.5,2)
cols = seq_along(ts)
for(i in seq_along(ts)){
t=ts[i]
lines(x,exp(-t/x),col=cols[i])
}
legend(x='topright',
title='tf-idf value',
legend=ts,
lty=1,
col=cols,
bty='n')
另一个方面是运行时间。 quickMarkers 作者运行了大约3秒钟,而运行 edgeR 则花费了大约30分钟(假如你尝试一下DEseq2,会奔溃哦)。其他标记物查找方法比这更快,但通常属于“每次比较几分钟”组,而不是“基本瞬时”。这看似微不足道,但这意味着可以自由地快速尝试许多不同的比较,这通常会非常有用。
这并不是说目前流行的包执行的差异表达对单细胞数据没有用处或不适用。
但作者希望在比较或设计单细胞数据的差异表达时,将基因的这一特性量化为非常特定于正在考虑的簇/细胞类型。在处理单细胞数据时,这是一个有用的区别,作者希望其他人也会这样做。