去年我在《生信技能树》给学员分享过一个案例:GSE16561 复现,小众的illumina bead芯片,链接在:http://www.bio-info-trainee.com/7483.html ,当时大费周折才拿到跟其数据集原始文献类似的结果,已经是很满意了。最近把这个数据集作为任务安排给最新学徒们,他们反馈给我的结果让我丈二和尚摸不着头脑,居然是百分百还原文献结果,如下所示的差异基因列表:

那我就纳闷了,赶紧检查他附送的代码 。
第一步:拿到表达矩阵
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
# 载入包
library(AnnoProbe)
library(GEOquery)
gset <- geoChina("GSE16561")
load('GSE16561_eSet.Rdata')
a=gset[[1]] #
dat=exprs(a) #a现在是一个对象,取a这个对象通过看说明书知道要用exprs这个函数
dim(dat)#看一下dat这个矩阵的维度
dat[1:4,1:4] #查看dat这个矩阵的1至4行和1至4列,逗号前为行,逗号后为列
boxplot(dat[,1:5],las=2)
# 可以看到box-and-whisker plot 的box基本上差不多
# 使用limma包进行归一化 数据集的数据便有了相似的分布
library(limma)
dat=normalizeBetweenArrays(dat)
boxplot(dat[,1:5],las=2)
#通过查看说明书知道取对象a里的临床信息用pData
pd=pData(a)
## 挑选一些感兴趣的临床表型。
library(stringr)
## 结果是个包含'Stroke','Control'的向量
group_list=ifelse(grepl('Stroke',pd$title),'Stroke','Control')
## the counts at each combination
table(group_list)
真的是诡异,我看了看,它这个表达矩阵,起初呢,表达量很明显是被zscore了,所以值是围绕着0上下起伏。然后他自作聪明使用了limma包附带的normalizeBetweenArrays函数,如下所示:
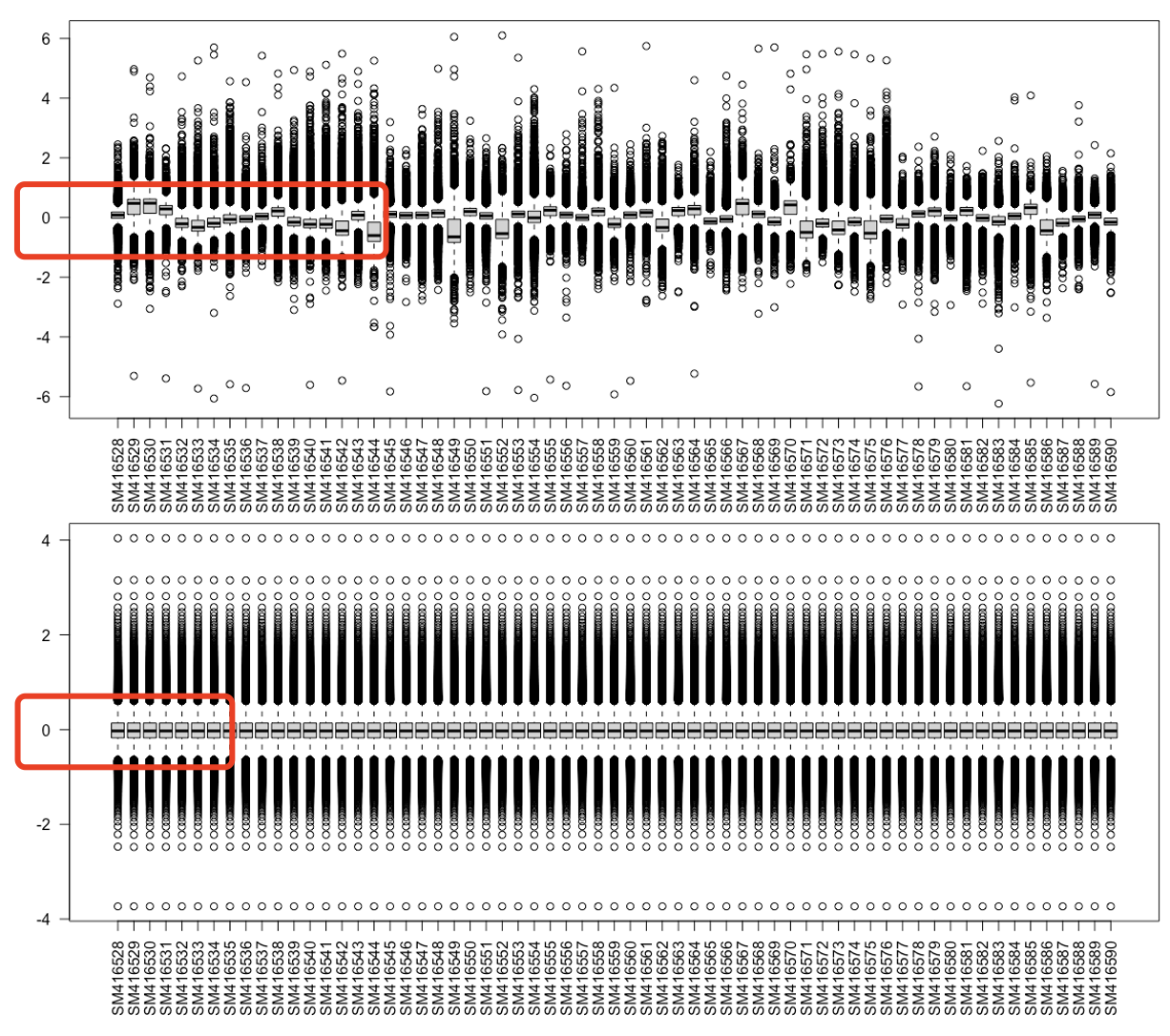
后面的差异分析,居然,他完全就是拿这个被zscore而且normalizeBetweenArrays函数处理的矩阵,我嘞个去,这么多细节,他全部做错了,但是它最后得到的差异基因反而和文章是几乎一模一样,包括挑出来的基因进行热图可视化,如下:

如果你仔细看两个表达矩阵的PCA,你甚至完全看不出来:
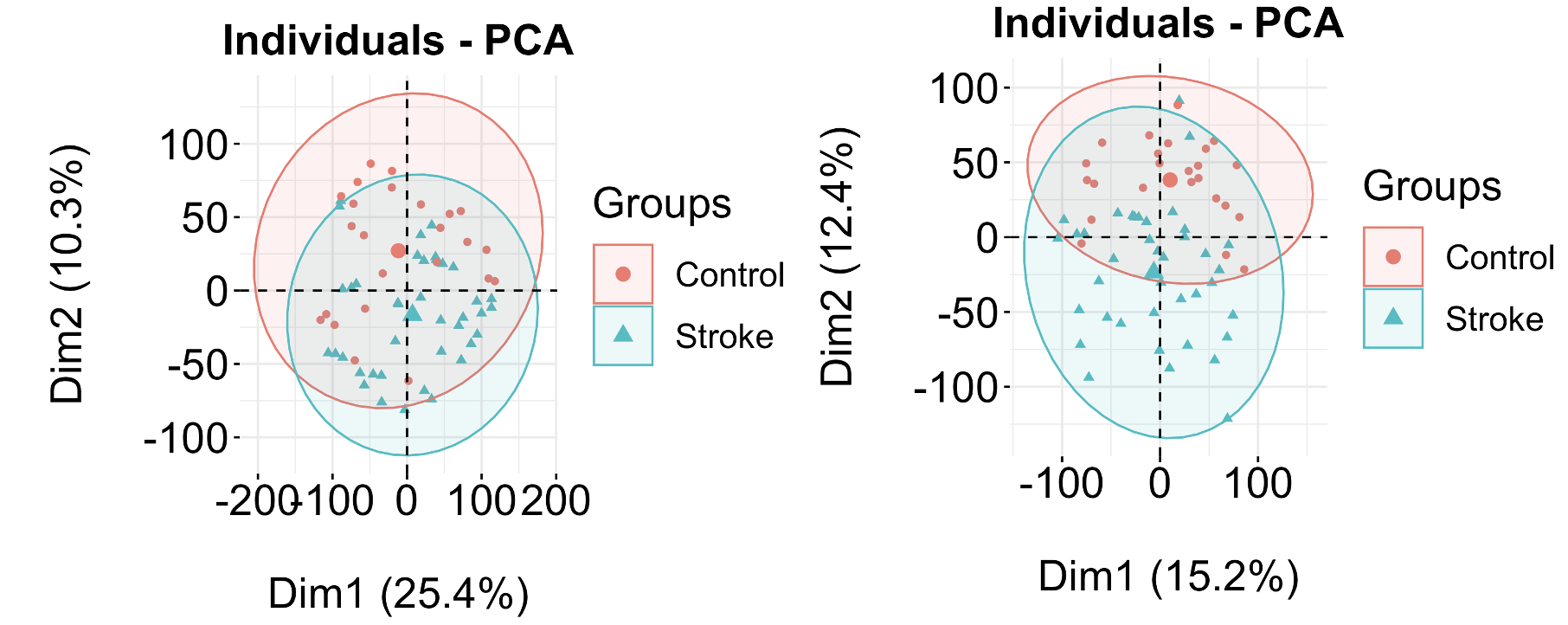
那么,我之前给出来的正确的答案的表达矩阵如何得到的呢,代码如下:
rm(list = ls())
# https://ftp.ncbi.nlm.nih.gov/geo/series/GSE16nnn/GSE16561/suppl/GSE16561_RAW.txt.gz
b=read.table('GSE16561_RAW.txt',header = T)
b[1:4,1:4]
rownames(b)=b[,1]
b=b[,-1]
boxplot(b[,1:5],las=2)
b=log(b+1)
boxplot(b[,1:5],las=2)
dat=b
library(limma)
dat=normalizeBetweenArrays(dat)
boxplot(dat,las=2)
pd=pData(a)
colnames(dat)
library(stringr)
group_list= str_split(colnames(dat) ,'_',simplify = T)[,2]
table(group_list)
也就是说,我并没有使用getGEO函数下载到的表达矩阵,因为安装我的经验,根据boxplot判断,这个表达矩阵已经被zscore了,理论上没办法进行后续分析了。
现实狠狠的扇了我一巴掌,哪怕表达矩阵已经被zscore了,居然拿到的差异基因没有问题。本来呢,我是怀疑这个数据集本身的文献,2011发表的那个就错了。
接下来我就比较了两次差异分析的区别:
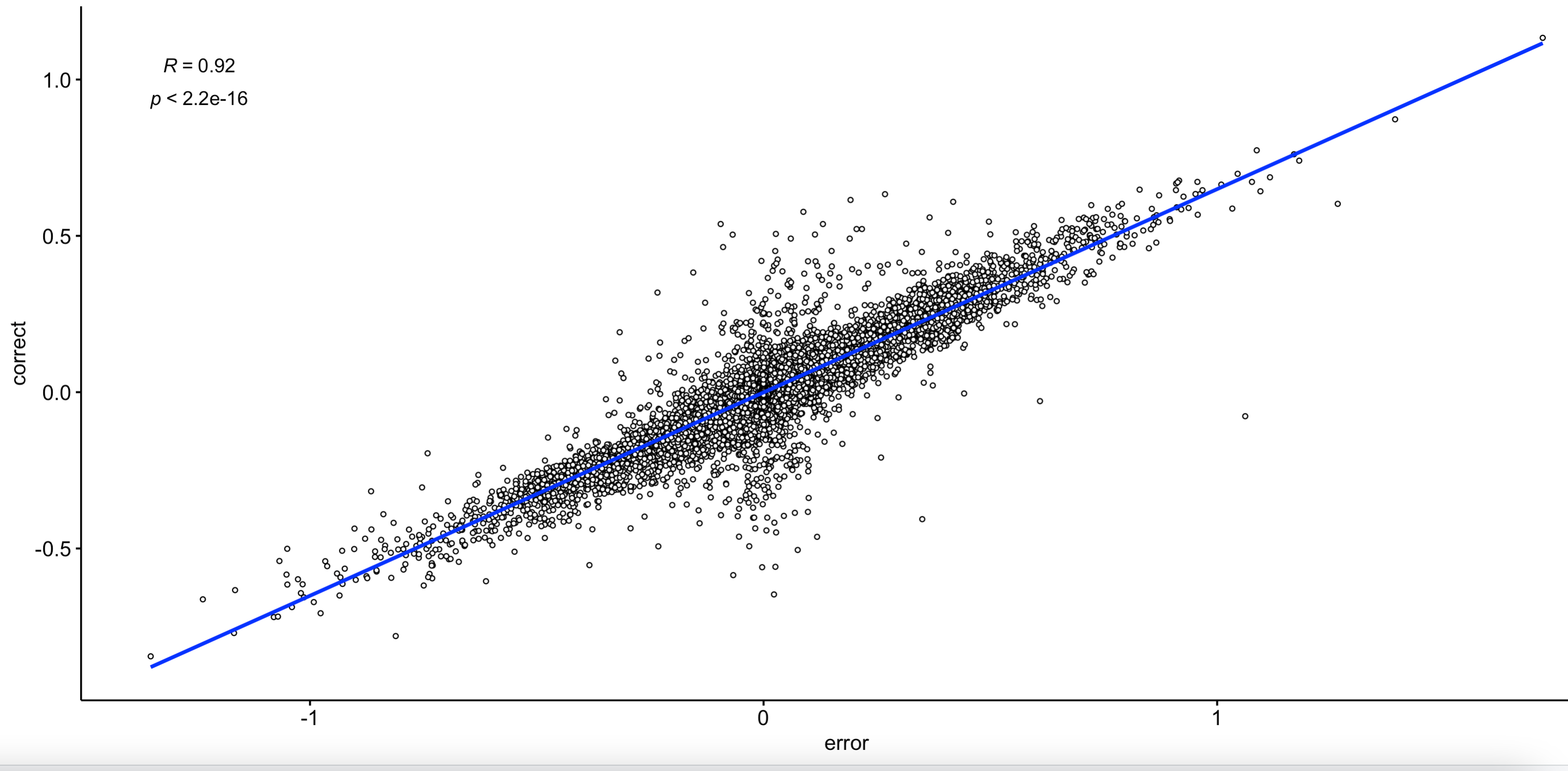
这个差异基本上可以忽略了。
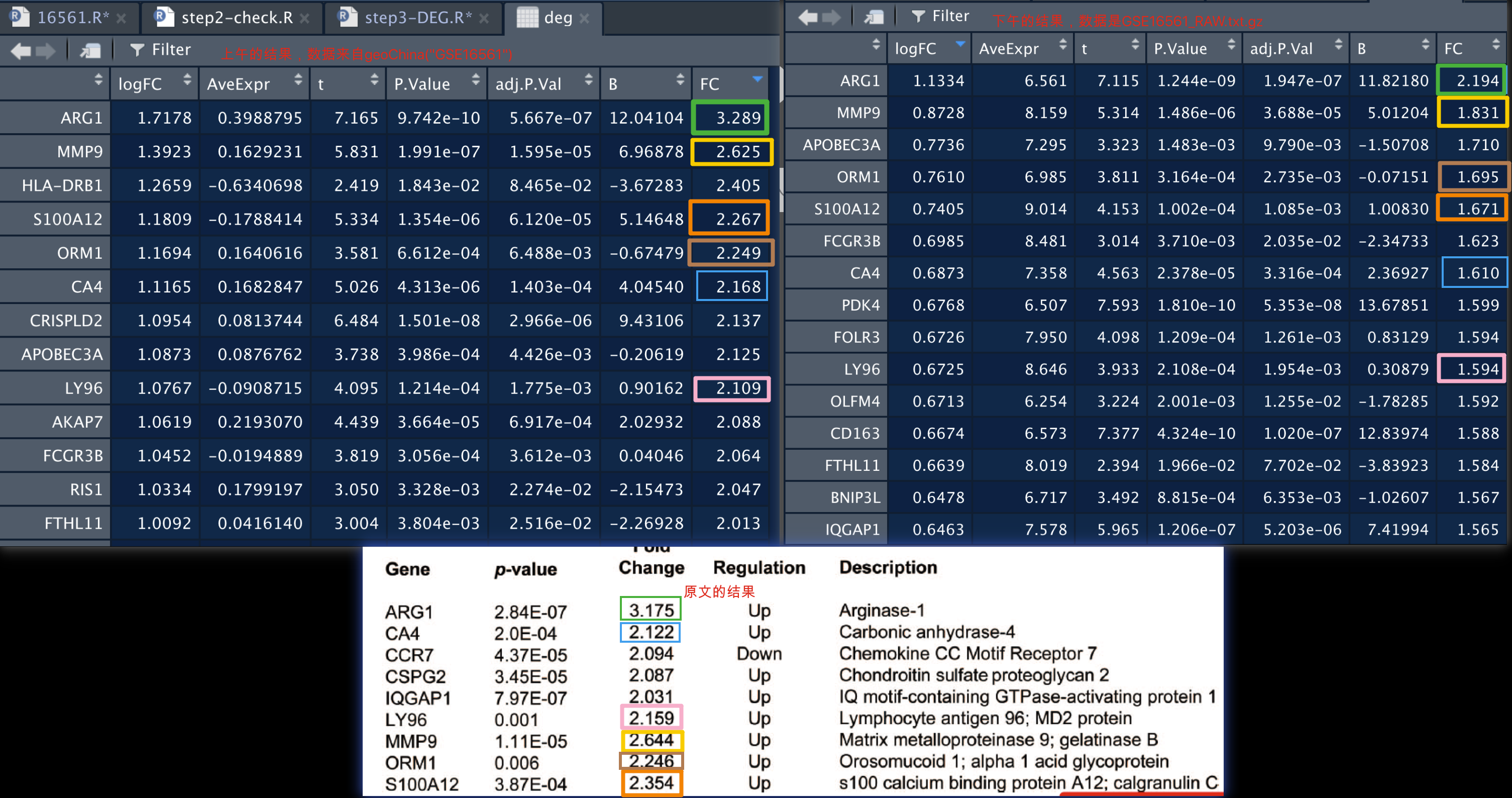
可以看到,上下调基因大体上是一致的,至于数值上的差异,本来就不重要。成比例的变动,等价于没有变化。
但是,背后的统计学原理我还没有想清楚,为什么表达矩阵已经被zscore了却并不是非常的影响它的后续差异分析呢?