前面我们的明码标价之普通转录组上游分析,受到了各大热心粉丝的吐槽,觉得太简单了我们居然还好意思收费。
额,其实呢,这些粉丝应该是“饱汉不知饿汉饥”,其实数据分析这个技能啊,难者不会,会者不难。而且普通转录组上游分析仅仅是一个引子,又不是说我们只能提供这些粗浅的分析,转录组相关的绝大部分高级分析我们都是有现成的代码,而且都分享过一些教程:
- 用LeafCutter探索转录组数据的可变剪切,需要star软件的
- 使用SGSeq探索可变剪切,一个读取bam文件的R包
最近看到了一个比较新,而且发表的杂志也还不错的工具:SUPPA2

首先认识 SUPPA 软件,它有5个子命令,分别如下:
- generateEvents: Generates events from an annotation.
- psiPerEvent : Quantifies event inclusion levels (PSIs) from multiple samples.
- psiPerIsoform : Quantifies isoform inclusion levels (PSIs) from multiple samples.
- diffSplice : Calculate differential splicing across multiple conditions with replicates.
- clusterEvents : Cluster events according to PSI values across conditions.
可以根据已知的 gtf文件信息针对多种可变剪切形式进行差异分析,它还提供了一些独立的py脚本文件,如下所示:

准备工作
包括conda安装,以及SUPPA2环境安装,参考转录组fasta序列获取和构建索引文件,参考gtf文件处理。
统一使用gencode最新版: https://www.gencodegenes.org/human/
mkdir -p SUPPA2/ref
cd SUPPA2/ref
nohup wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_37/gencode.v37.annotation.gtf.gz &
nohup wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_37/gencode.v37.transcripts.fa.gz &
gunzip gencode.v37.annotation.gtf.gz
gunzip gencode.v37.transcripts.fa.gz
# 1.4G 2月 17 16:50 gencode.v37.annotation.gtf
使用conda创建rna小环境:
代码如下:
conda create -n rna
conda activate rna
conda install -y trim-galore
# python-3.7.9 | 57.3 MB, openjdk-10.0.2 | 189.2 MB
conda install -y star hisat2 bowtie2 # 没有其它依赖
conda install -y subread bedtools deeptools
conda install -y salmon=1.4.0
conda install -y -c bioconda suppa
下载参考转录组fasta序列文件然后使用salmon构建index文件
conda activate rna
nohup salmon index -t gencode.v37.transcripts.fa -i gencode.v37.transcripts.salmon.index &
得到的salmon的index是一个文件夹,内容如下:
$ ls -lh gencode.v37.transcripts.salmon.index/|cut -d" " -f 5-
913K 2月 17 17:19 complete_ref_lens.bin
95M 2月 17 17:23 ctable.bin
4.3M 2月 17 17:23 ctg_offsets.bin
175K 2月 17 17:19 duplicate_clusters.tsv
1.1K 2月 17 17:24 info.json
90M 2月 17 17:24 mphf.bin
479M 2月 17 17:24 pos.bin
496 2月 17 17:24 pre_indexing.log
23M 2月 17 17:23 rank.bin
1.8M 2月 17 17:23 refAccumLengths.bin
16K 2月 17 17:24 ref_indexing.log
913K 2月 17 17:23 reflengths.bin
92M 2月 17 17:23 refseq.bin
45M 2月 17 17:23 seq.bin
126 2月 17 17:24 versionInfo.json
下载基因注释gtf文件然后使用suppa构建可变剪切事件综合文件
统一使用gencode最新版: https://www.gencodegenes.org/human/
对gtf文件的全部转录本走 suppa.py generateEvents 命令:
# 使用 generateEvents 命令根据基因组的gtf注释文件生成所有的可变剪切事件,格式保存为ioe格式
# 针对 SE SS MX RI FL 可变剪切批量分析的
suppa.py generateEvents \
-i gencode.v37.annotation.gtf \
-o gencode.v37.events -e SE SS MX RI FL -f ioe
得到的文件如下:
$ wc -l *ioe
20838 gencode.v37.events_A3_strict.ioe
18921 gencode.v37.events_A5_strict.ioe
112101 gencode.v37.events_AF_strict.ioe
37317 gencode.v37.events_AL_strict.ioe
8629 gencode.v37.events_MX_strict.ioe
8474 gencode.v37.events_RI_strict.ioe
50562 gencode.v37.events_SE_strict.ioe
可以看到是,基于 gencode.v37.annotation.gtf 文件的7种全部的可能的可变剪切事件,都被列出来了。
# 合并所有的ioe文件
awk 'FNR==1 && NR!=1 { while (/^<header>/) getline; } 1 {print}' *.ioe > gencode.v37.all.events.ioe
wc -l gencode.v37.all.events.ioe
# 256842 gencode.v37.all.events.ioe
上游分析拿到tpm矩阵
测序数据获取
得到文件如下:
ls -lh rawdata/*gz|cut -d" " -f 5-
1.7G 2月 8 14:33 rawdata/SRR1513329_1.fastq.gz
1.7G 2月 8 14:33 rawdata/SRR1513329_2.fastq.gz
1.7G 2月 8 14:33 rawdata/SRR1513330_1.fastq.gz
1.7G 2月 8 14:33 rawdata/SRR1513330_2.fastq.gz
1.8G 2月 8 14:33 rawdata/SRR1513331_1.fastq.gz
1.8G 2月 8 14:33 rawdata/SRR1513331_2.fastq.gz
1.8G 2月 8 14:33 rawdata/SRR1513332_1.fastq.gz
1.8G 2月 8 14:33 rawdata/SRR1513332_2.fastq.gz
1.9G 2月 8 14:33 rawdata/SRR1513333_1.fastq.gz
2.0G 2月 8 14:33 rawdata/SRR1513333_2.fastq.gz
1.7G 2月 8 14:33 rawdata/SRR1513334_1.fastq.gz
1.8G 2月 8 14:33 rawdata/SRR1513334_2.fastq.gz
然后质控,走fastqc软件,查看报告
fastqc -t 2 *.fastq.gz -o ../qc
通常情况下大家需要针对clean后的fq文件进行后续的salmon流程定量获得各个样品的TPM值 。
走trim流程
因为是双端测序,所以:
cd rawData
ls *gz|cut -d"_" -f 1|sort -u |while read id;do
nohup trim_galore -q 25 --phred33 --length 36 --stringency 3 --paired -o ../cleanData ${id}*.gz &
done
得到的clean的fq文件如下:
$ ls -lh *gz|cut -d" " -f 5-
1.6G 2月 18 10:00 SRR1513329_1_val_1.fq.gz
1.6G 2月 18 10:00 SRR1513329_2_val_2.fq.gz
1.6G 2月 18 10:00 SRR1513330_1_val_1.fq.gz
1.6G 2月 18 10:00 SRR1513330_2_val_2.fq.gz
1.7G 2月 18 10:02 SRR1513331_1_val_1.fq.gz
1.8G 2月 18 10:02 SRR1513331_2_val_2.fq.gz
1.7G 2月 18 10:02 SRR1513332_1_val_1.fq.gz
1.7G 2月 18 10:02 SRR1513332_2_val_2.fq.gz
1.9G 2月 18 11:15 SRR1513333_1_val_1.fq.gz
1.9G 2月 18 11:15 SRR1513333_2_val_2.fq.gz
1.7G 2月 18 18:04 SRR1513334_1_val_1.fq.gz
1.7G 2月 18 18:04 SRR1513334_2_val_2.fq.gz
项目介绍 分组情况如下:
negative control siRNA :SRR1513329,SRR1513330,SRR1513331
TRA2A/B siRNA :SRR1513332,SRR1513333,SRR1513334
salmon流程定量获得各个样品的TPM值
## 定量获得TPM值
mkdir salmon_output
cd cleanData
index=../ref/gencode.v37.transcripts.salmon.index/
ls *gz|cut -d"_" -f 1|sort -u |while read id;do
nohup salmon quant -i $index -l ISF --gcBias \
-1 ${id}_1_val_1.fq.gz -2 ${id}_2_val_2.fq.gz -p 2 \
-o ../salmon_output/${id}_output 1>${id}_salmon.log 2>&1 &
done
## quant.sf文件很重要,要用于后续的分析
##ENST和ENSG的前三个字母(ENS),意思是“ENSENMBLE”。T是指转录本。G是指基因。当然,也会看到ENSP,P自然是指蛋白质了。
几分钟就可以走完全部的salmon流程,每一个样品虽然说会输出一个文件夹,但是最重要的都是各自的 quant.sf文件,行数都是统一的:
$ wc -l ../salmon_output/*/quant.sf
233653 ../salmon_output/SRR1513329_output/quant.sf
233653 ../salmon_output/SRR1513330_output/quant.sf
233653 ../salmon_output/SRR1513331_output/quant.sf
233653 ../salmon_output/SRR1513332_output/quant.sf
233653 ../salmon_output/SRR1513333_output/quant.sf
233653 ../salmon_output/SRR1513334_output/quant.sf
各自的 quant.sf文件内容如下:
==> ../salmon_output/SRR1513334_output/quant.sf <==
Name Length EffectiveLength TPM NumReads
ENST00000456328.2|ENSG00000223972.5|OTTHUMG00000000961.2|OTTHUMT00000362751.1|DDX11L1-202|DDX11L1|1657|processed_transcript| 1657 1587.157 0.000000 0.000
ENST00000450305.2|ENSG00000223972.5|OTTHUMG00000000961.2|OTTHUMT00000002844.2|DDX11L1-201|DDX11L1|632|transcribed_unprocessed_pseudogene| 632 452.000 0.000000 0.000
ENST00000488147.1|ENSG00000227232.5|OTTHUMG00000000958.1|OTTHUMT00000002839.1|WASH7P-201|WASH7P|1351|unprocessed_pseudogene| 1351 1273.856 3.098744 27.492
tpm表达量矩阵的获取
这个时候,可以直接使用作者的 multipleFieldSelection.py 脚本,对我们的全部样品的 quant.sf文件 进行信息提取即可。其实自己写一个脚本也是很简单的:
-kindicates the row used as index-fTPM值在第四列,在这里要提取每个样本的TPM值
## 提取所有样本的TPM值并合并为一个文件
cd ../salmon_output/
multipleFieldSelection.py \
-i SRR*/quant.sf -k 1 -f 4 \
-o iso_tpm.txt
得到 iso_tpm.txt的内容 ,还需要格式化一下,因为它跟gtf文件里面的基因信息不一致。
# gencode.v37.transcripts.fa 内容如下
>ENST00000450305.2|ENSG00000223972.5|OTTHUMG00000000961.2|OTTHUMT00000002844.2|DDX11L1-201|DDX11L1|632|transcribed_unprocessed_pseudogene|
# gencode.v37.annotation.gtf 内容如下
chr1 HAVANA gene 11869 14409 . + . gene_id "ENSG00000223972.5"; gene_type "transcribed_unprocessed_pseudogene"; gene_name "DDX11L1"; level 2; hgnc_id "HGNC:37102"; havana_gene "OTTHUMG00000000961.2";
各自的 quant.sf文件里面的ID是gencode.v37.transcripts.fa里面的转录本ID,有点过于复杂了,所以需要进行简化,需要与 gencode.v37.annotation.gtf 里面的转录本ID保持一致。
perl -alne '{/(\|.*\|)\t/; ;s/$1//g;s/\|//g;print}' iso_tpm.txt > iso_tpm_formatted.txt
输出文件是 iso_tpm_formatted.txt ,是 233653 行的普通文本,可以看到简洁很多:
SRR1513329_output SRR1513330_output SRR1513331_output SRR1513332_output SRR1513333_output SRR1513334_output
ENST00000456328.2 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
ENST00000450305.2 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
拿到了这样的以转录本ID为行的tpm矩阵,再加上 gencode.v37.annotation.gtf 的可变剪切形式文件,就可以进行下游分析获取统计学显著的可变剪切事件。
下游分析获取统计学显著的可变剪切事件
项目介绍 分组情况如下:
negative control siRNA :SRR1513329,SRR1513330,SRR1513331
TRA2A/B siRNA :SRR1513332,SRR1513333,SRR1513334
对tpm表达量矩阵,根据可变剪切事件计算psi值矩阵:
ioe_merge_file=../ref/gencode.v37.all.events.ioe
ls $ioe_merge_file
suppa.py psiPerEvent \
-i $ioe_merge_file -e iso_tpm_formatted.txt -o project_events 1>psiPerEvent_log.txt 2>&1 &
# 会输出 project_events.psi 文件
同时可以检查看看 psiPerEvent_log.txt 文件。
project_events.psi 文件,内容如下所示:
SRR1513329_output SRR1513330_output SRR1513331_output SRR1513332_output SRR1513333_output SRR1513334_output
ENSG00000000003.15;A5:chrX:100635746-100636191:100635746-100636608:- 0.20449107275352194 0.20506877462332357 0.18431225084229827 0.32174184236220055 0.27519806289263166 0.318119876931685
ENSG00000000003.15;A5:chrX:100635746-100636608:100635746-100636793:- 1.0 1.0 0.9009096685944438 1.0 0.9145937251391483 1.0
根据分组做差异可变剪切事件寻找
把前面的全部转录本可变剪切事件的psi文件以及 iso_tpm_formatted.txt ,根据分组信息,拆分为两个psi矩阵 和两个tpm矩阵。
# negative control siRNA :SRR1513329,SRR1513330,SRR1513331
cut -f 1-4 project_events.psi > control.psi
cut -f 1-4 iso_tpm_formatted.txt > control.tpm
# TRA2A/B siRNA :SRR1513332,SRR1513333,SRR1513334
cut -f 1,5-7 project_events.psi > treat.psi
cut -f 1,5-7 iso_tpm_formatted.txt > treat.tpm
# 很有意思的事情是,这个时候的表头似乎是并不是很重要
然后走 suppa.py diffSplice 命令,对两个分组的psi矩阵 和tpm矩阵进行 差异可变剪切事件寻找
ioe_merge_file=../ref/gencode.v37.all.events.ioe
ls $ioe_merge_file
suppa.py diffSplice \
-m empirical -gc -i $ioe_merge_file \
--save_tpm_events \
-p treat.psi control.psi \
-e treat.tpm control.tpm \
-o project_diffSplice # 前缀
差异分析生成下列3个文件:
ls -lh project_diffSplice* |cut -d" " -f 5-
7.9M 2月 18 09:16 project_diffSplice_avglogtpm.tab
23M 2月 18 09:38 project_diffSplice.dpsi
29M 2月 18 09:38 project_diffSplice.psivec
到此为止,统计学显著的可变剪切事件就获取完毕,后面就是各式各样的可视化啦!
当然了,一般来说,统计学显著的才有意义:
$ cat project_diffSplice.dpsi|perl -alne '{print if $F[2] <0.01}' |wc
118 353 11552
$ cat project_diffSplice.dpsi|perl -alne '{print if $F[2] <0.05}' |wc
750 2249 74204
查看差异比较大的可变剪切事件:
treat-control_dPSI treat-control_p-val
ENSG00000008517.18;AF:chr16:3065297:3065494-3065784:3065677:3065710-3065784:+ -1.0 0.0
ENSG00000009307.16;SE:chr1:114738072-114749821:114750207-114757925:- 0.6712142721 0.008991009
ENSG00000009307.16;SE:chr1:114739890-114749821:114750207-114757925:- 0.1488639077 0.008991009
这些差异比较大的可变剪切事件可以进行一些可视化,可以是igv里面的,举例如下:
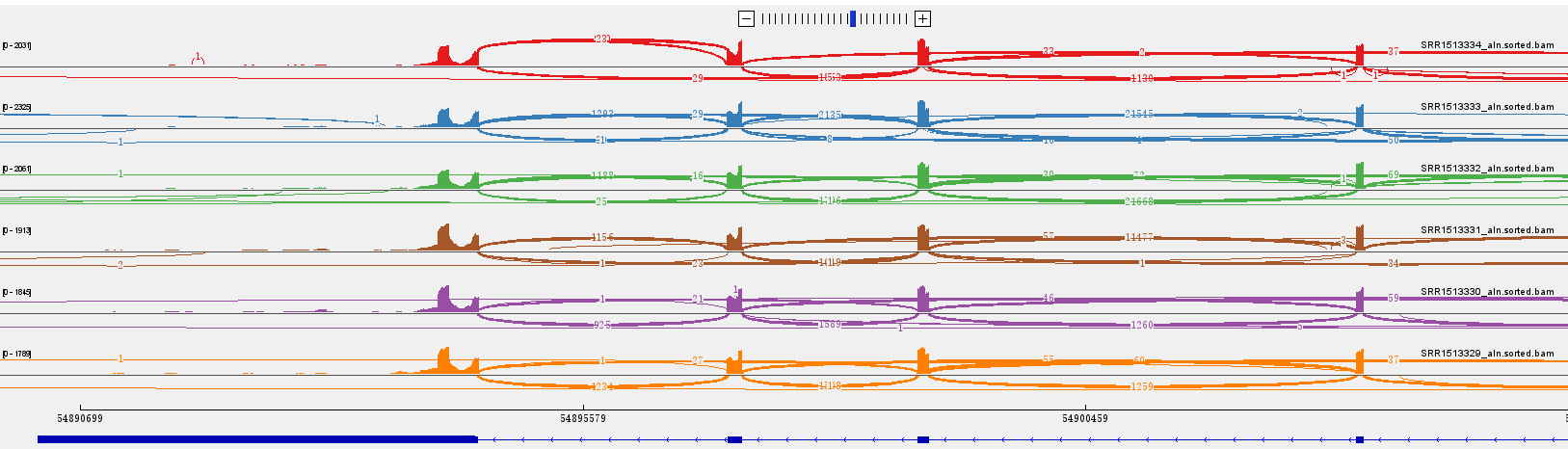
网页工具绝对是完成不了这样的命令行数据分析哦
这个是基于Linux的ngs数据的上游处理,目前没有成熟的网页工具支持这样的分析。其实呢,如果你有时间请务必学习编程基础,自由自在的探索海量的公共数据,辅助你的科研,那么:
如果你没有时间从头开始学编程,也可以委托专业的团队付费拿到同样的数据分析, 这样的20个样品以内的简单分组转录组测序数据的表达矩阵获取,首先需800元,然后如果要进行高级分析,比如上面讲解的可变剪切,再需要800元,仍然是可以拿到全部的数据和代码哦!
- 需要自己读文献筛选合适的数据集
- 提供半个小时左右的一对一讲解转录组数据处理背景知识。
如果需要委托,直接在我们《生信技能树》公众号留言即可,我们会安排合适的生信工程师对接具体的项目。