前面我们公布了《cytof数据资源介绍(文末有交流群)》,现在就开始正式手把手教学。
上一讲我们构造好了SingleCellExperiment对象,后续全部的分析都会以这个SingleCellExperiment对象为准,大家务必熟悉SingleCellExperiment对象的各种结构,教程见:cytofWorkflow之构建SingleCellExperiment对象(二)。有了这个SingleCellExperiment对象,并且成功分成了不同的生物学亚群,就可以看看不同组样本的不同生物学亚群的比例差异。
首先从SingleCellExperiment对象可以计算不同细胞亚群的比例
前面的教程:cytofWorkflow之聚类分群(四),每个病人的20个亚群细胞比例都展现出来了,这个时候可以隐隐约约看到某一些细胞亚群在某一个组特异性的占比较高。
plotAbundances(sce, k = "meta20", by = "sample_id")
ggsave2(filename = paste0(pro,'_plotAbundances_barplot.pdf'))
plotAbundances(sce, k = "meta20", by = "cluster_id", shape_by = "patient_id")
ggsave2(filename = paste0(pro,'_plotAbundances_boxplot.pdf'))
我们其实可以自己写函数计算这个比例表格,自行绘图。代码如下:
rm(list = ls())
suppressPackageStartupMessages(library(flowCore))
suppressPackageStartupMessages(library(diffcyt))
suppressPackageStartupMessages(library(HDCytoData))
require(cytofWorkflow)
load(file = 'K20_output_of_cytofWorkflow.Rdata')
ns <- table(cluster_id = cluster_ids(sce, 'meta20'),
sample_id = sample_ids(sce))
fq <- prop.table(ns, 2) * 100
df <- as.data.frame(fq)
library(reshape2)
head(df)
dat=dcast(df,cluster_id~sample_id)
得到的结果表格如下:
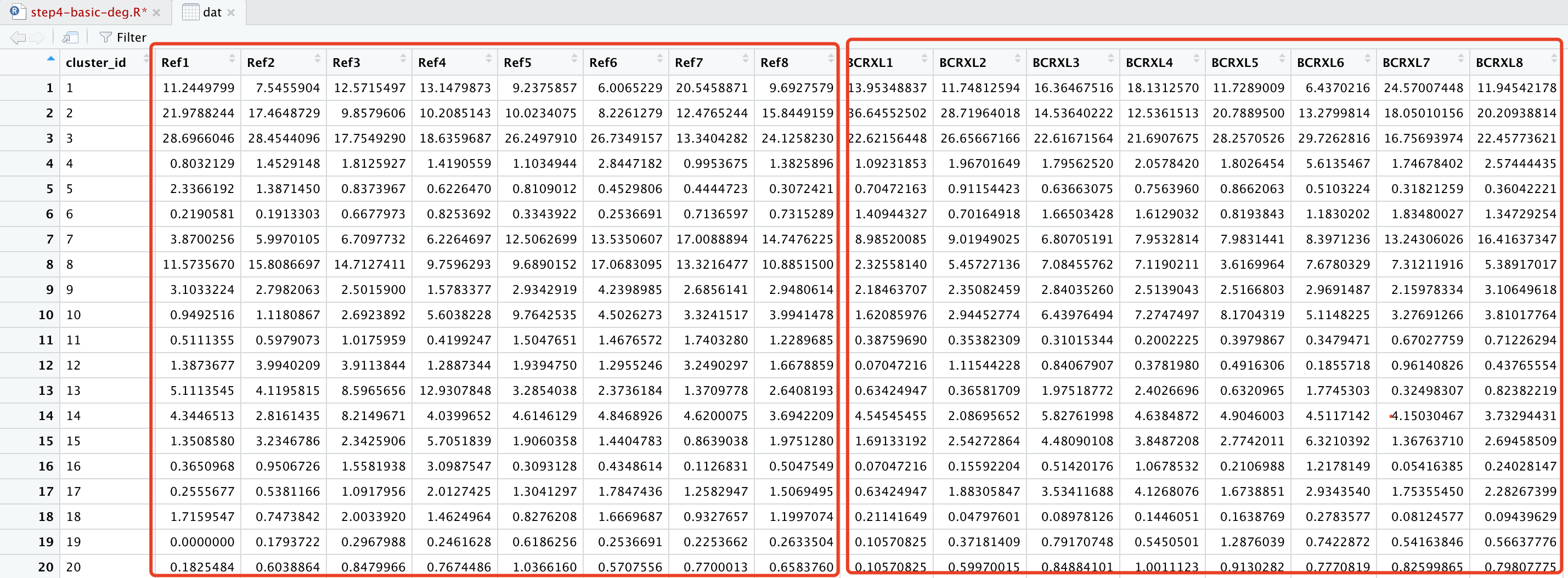
通常呢,这个表格需要写出成为csv文件后给跟你合作的生物学家。
然后对不同细胞亚群的比例表格进行差异分析
首先是简单的T检验
普通的差异分析,其实简单的t检验也是可以的。
ei <- metadata(sce)$experiment_info
ei
dat$p=apply(dat,1,function(x){
t.test(as.numeric(x[-1])~ei$condition)$p.value
})
dat$p
这样会发现, 符合要求的,有统计学显著变化的细胞亚群并不多。
然后使用R包进行差异分析 differential abundance (DA)
最常见的就是diffcyt包啦,Default = "diffcyt-DA-edgeR". 内置的统计学方法包括:
"diffcyt-DA-edgeR""diffcyt-DA-voom""diffcyt-DA-GLMM".
这个时候可以仅仅是考虑分组效应,也可以加入病人效应再进行差异分析,相当于转录组领域的去除批次效应。
(da_formula1 <- createFormula(ei,
cols_fixed = "condition",
cols_random = "sample_id"))
(da_formula2 <- createFormula(ei,
cols_fixed = "condition",
cols_random = c("sample_id", "patient_id")))
contrast <- createContrast(c(0, 1))
da_res1 <- diffcyt(sce,
formula = da_formula1, contrast = contrast,
analysis_type = "DA", method_DA = "diffcyt-DA-GLMM",
clustering_to_use = "meta20", verbose = FALSE)
da_res2 <- diffcyt(sce,
formula = da_formula2, contrast = contrast,
analysis_type = "DA", method_DA = "diffcyt-DA-GLMM",
clustering_to_use = "meta20", verbose = FALSE)
names(da_res1)
rowData(da_res1$res)
FDR_cutoff=0.05
table(rowData(da_res1$res)$p_adj < FDR_cutoff)
table(rowData(da_res2$res)$p_adj < FDR_cutoff)
topTable(da_res2, show_props = TRUE, format_vals = TRUE, digits = 2)
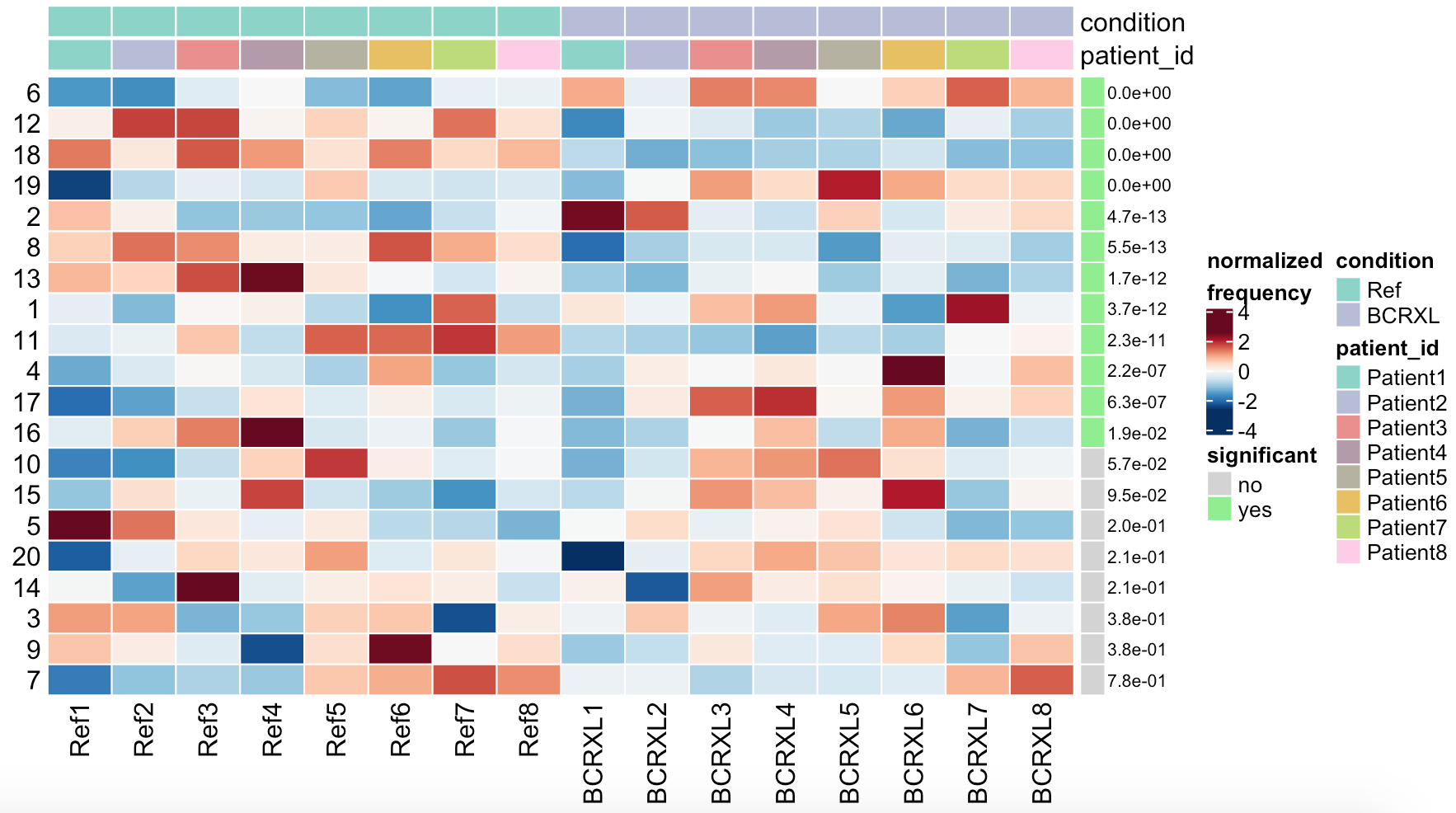
plotDiffHeatmap(sce, rowData(da_res2$res), all = TRUE, fdr = FDR_cutoff)
比较自己的t检验和R包差异分析结果
我们也可以拿到R包差异分析结果矩阵,代码如下:
df=topTable(da_res2, show_props = TRUE, format_vals = TRUE, digits = 2,show_all_cols=T)
df=as.data.frame(df)
df
write.csv(df,file = paste0(pro,'_dif.csv'))
可以看到:
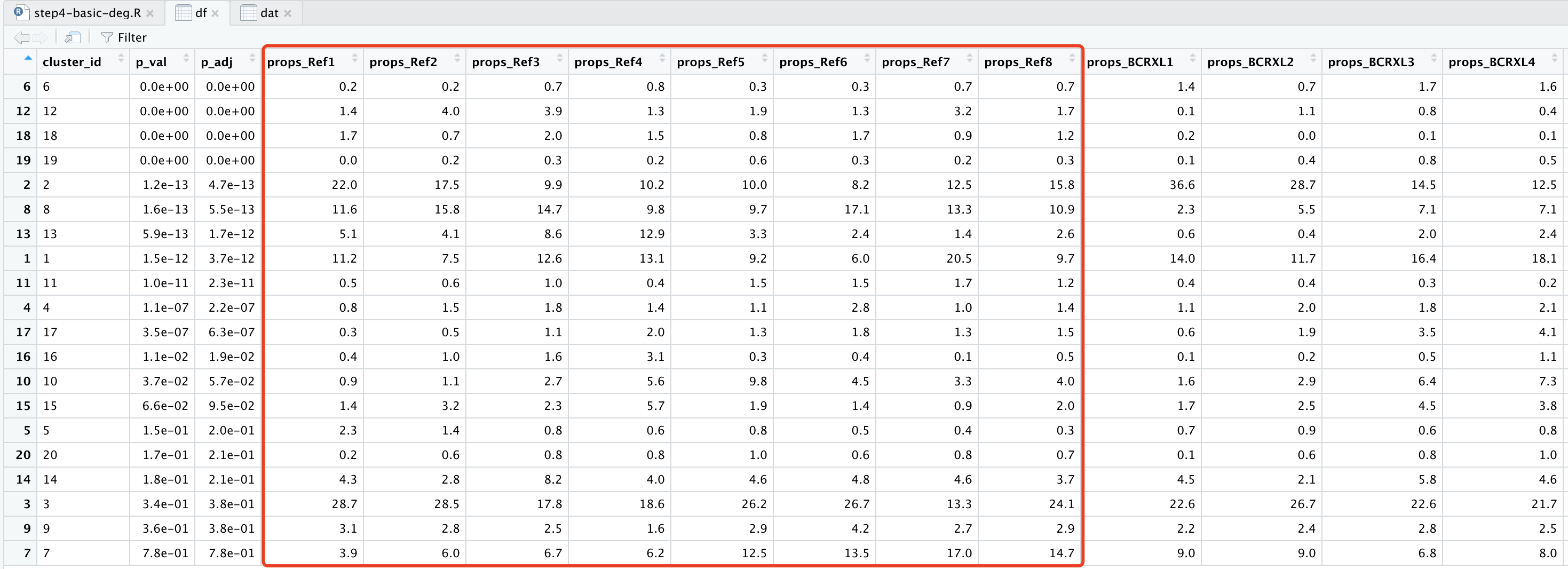
这20个细胞亚群在16个样品的比例矩阵都是一样的,就是计算P值很不一样哦。
当然,最后的结果也可以可视化,如下: