一般来说,大家拿到了感兴趣的基因集后,通常是做超几何分布检验看看富集到了什么生物学功能数据库,比如KEGG或者GO数据库,或者走gsea/gsva这样的富集分析,也是注释生物学功能数据库。 大家读我的表达芯片的公共数据库挖掘系列推文应该是够多了:
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
最近看到,单细胞领域有一个分析,可能是可以迁移到大家的常规表达量矩阵分析里面,就是最后大家拿到了基因集:
使用RcisTarget包进行转录因子富集分析
下载及安装就不多说了 ,加载好RcisTarget包后,可以查看其详细的文档:
# Explore tutorials in the web browser:
browseVignettes(package="RcisTarget")
vignette("RcisTarget") # open
核心就是 cisTarget()函数的使用,需要3个输入数据:
首先是配套数据库文件
不同物种不一样, 在 https://resources.aertslab.org/cistarget/ 查看自己的物种,按需下载,比如我这里就下载了人类和小鼠的数据:
# https://resources.aertslab.org/cistarget/
dbFiles <- c("https://resources.aertslab.org/cistarget/databases/homo_sapiens/hg19/refseq_r45/mc9nr/gene_based/hg19-500bp-upstream-7species.mc9nr.feather",
"https://resources.aertslab.org/cistarget/databases/homo_sapiens/hg19/refseq_r45/mc9nr/gene_based/hg19-tss-centered-10kb-7species.mc9nr.feather")
# dir.create("cisTarget_databases"); setwd("cisTarget_databases") # if needed
dbFiles <- c("https://resources.aertslab.org/cistarget/databases/mus_musculus/mm9/refseq_r45/mc9nr/gene_based/mm9-500bp-upstream-7species.mc9nr.feather",
"https://resources.aertslab.org/cistarget/databases/mus_musculus/mm9/refseq_r45/mc9nr/gene_based/mm9-tss-centered-10kb-7species.mc9nr.feather")
# mc9nr: Motif collection version 9: 24k motifs
dbFiles
for(featherURL in dbFiles)
{
download.file(featherURL, destfile=basename(featherURL)) # saved in current dir
# (1041.7 MB)
#
}
每个文件都是1G,下载好了存放在共享文件夹,可以多台电脑传输使用,没有必要每次都下载。
然后是基因集
一般来说呢,基因集可以是一次差异分析,挑选的符合统计学检验的,或者呢,直接挑选表达量或者离散度比较大的前500或者1000个基因,这个取决于大家的生物学研究目标。这里直接使用作者的 提供的:
library(RcisTarget)
# Load gene sets to analyze. e.g.:
geneList1 <- read.table(file.path(system.file('examples', package='RcisTarget'), "hypoxiaGeneSet.txt"),
stringsAsFactors=FALSE)[,1]
geneLists <- list(hypoxia=geneList1 )
geneLists
可以看到是 171个基因来自于hypoxiaGeneSet.txt文本文件,以symbol为名字:
> geneLists
$geneListName
[1] "ADM" "ADORA2B" "AHNAK2" "AK4" "AKAP12" "ALDOC"
[7] "ANG" "ANGPTL4" "ANKZF1" "ARTN" "ASPH" "ATF3"
[13] "ATG14" "ATXN1" "B3GNT4" "BBX" "BCOR" "BHLHE40"
[19] "BNIP3" "BNIP3L" "C12orf24" "C5orf13" "C7orf68" "CA9"
[25] "CADM1" "CAV1" "CCNG2" "CD59" "CITED2" "CSGALNACT1"
[31] "CSRP2" "CXCR4" "CYB5A" "CYP1B1" "CYR61" "DAAM1"
接着需要一个ID转换列表
也是直接使用RcisTarget包提供的即可
# Select motif database to use (i.e. organism and distance around TSS)
data(motifAnnotations_hgnc)
motifAnnotations_hgnc
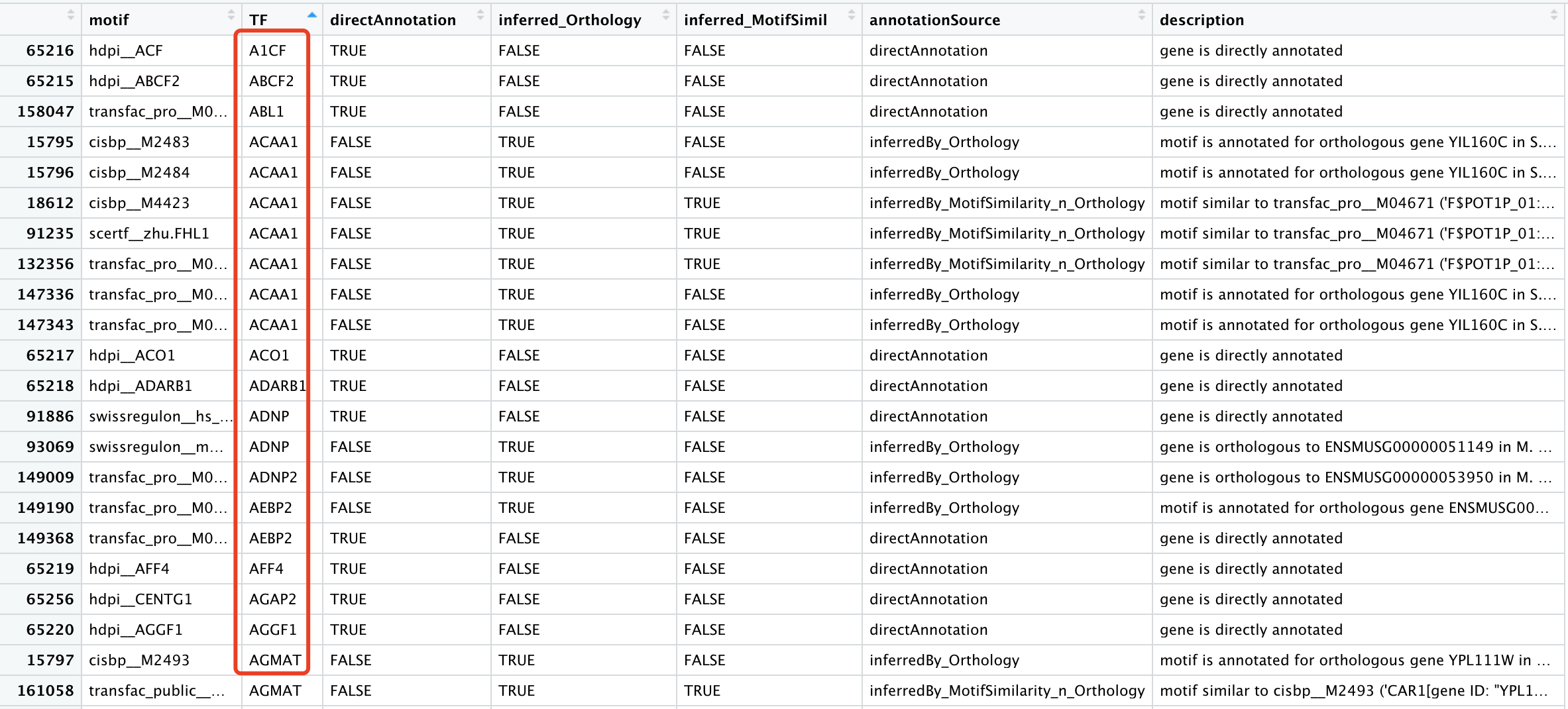
其中这个RcisTarget包内置的motifAnnotations_hgnc是16万行,可以看到每个基因有多个motif:
如果是其它物种,就需要自行去看文档,如何搞定这个ID转换列表啦。
最后就直接运行 cisTarget()函数
记住,前面我们下载好的 hg19-tss-centered-10kb-7species.mc9nr.feather 文件,需要存储在 当前工作目录文件夹cisTarget_databases里面哦:
motifRankings <- importRankings("cisTarget_databases/hg19-tss-centered-10kb-7species.mc9nr.feather")
# Motif enrichment analysis:
motifEnrichmentTable_wGenes <- cisTarget(geneLists, motifRankings,
motifAnnot=motifAnnotations_hgnc)
这个motifRankings是读取了数据库文件后的S4对象,最重要的信息是这个S4对象里面的rankings表格,节选如下:

解析结果
最后得到的结果是motifEnrichmentTable_wGenes,一个简单的数据框,总共是100个motif被富集到了,节选如下:
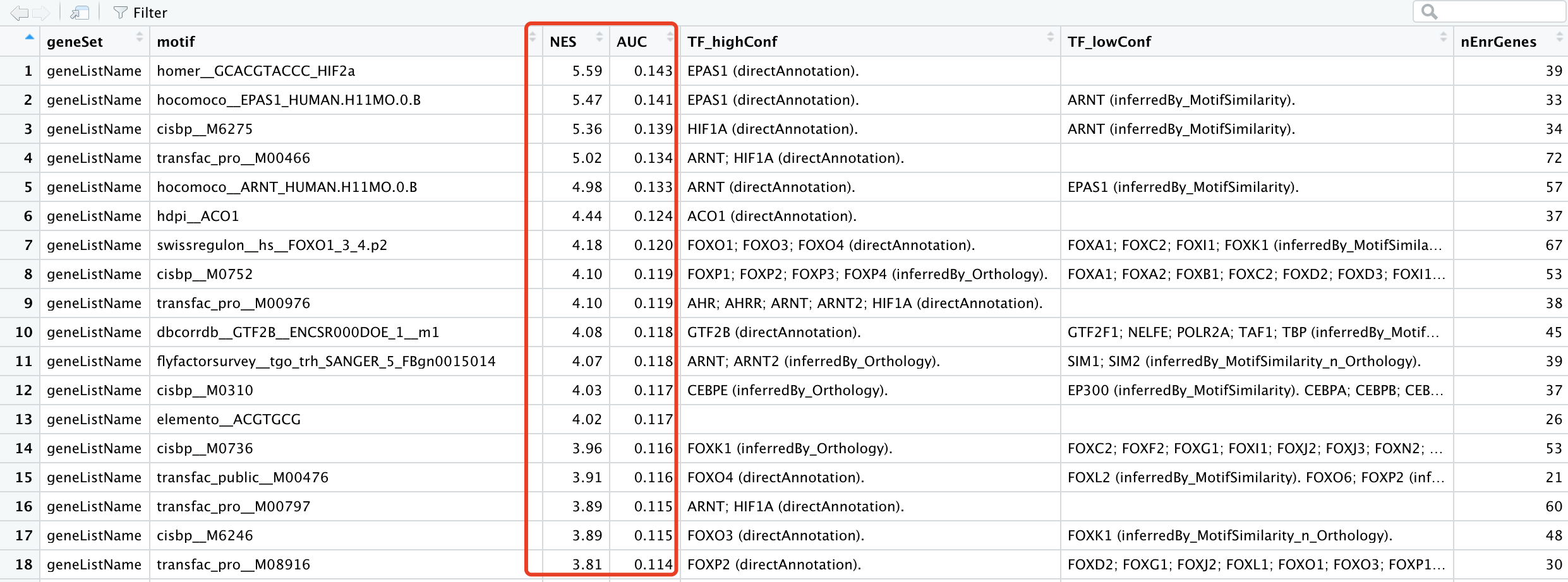
可以看到,NES概念大家都不陌生了,就是GSEA分析的结果而已,AUC的概念比较新颖。
其中EPAS1 (directAnnotation)这个基因命中了两个转录因子:
hocomoco__EPAS1_HUMAN.H11MO.0.B
homer__GCACGTACCC_HIF2a
可以去前面的数据框查询看看,首先在RcisTarget包内置的motifAnnotations_hgnc,16万行数据信息里面可以找寻到这两个转录因子。
然后是在motifRankings查看:
motifs=unlist(as.data.frame(motifRankings@rankings[,1]))
match('homer__GCACGTACCC_HIF2a',motifs)
match('hocomoco__EPAS1_HUMAN.H11MO.0.B',motifs)
因为通过RcisTarget包的 cisTarget()函数,一句代码完成的转录因子富集分析,等价于下面的3个步骤。
# 1. Calculate AUC
motifs_AUC <- calcAUC(geneLists, motifRankings)
# 2. Select significant motifs, add TF annotation & format as table
motifEnrichmentTable <- addMotifAnnotation(motifs_AUC,
motifAnnot=motifAnnotations_hgnc)
# 3. Identify significant genes for each motif
# (i.e. genes from the gene set in the top of the ranking)
# Note: Method 'iCisTarget' instead of 'aprox' is more accurate, but slower
motifEnrichmentTable_wGenes <- addSignificantGenes(motifEnrichmentTable,
geneSets=geneLists,
rankings=motifRankings,
nCores=1,
method="aprox")
后面我们再具体讲解,这3个步骤是如何挑选到这100个motif的。