学徒培养进行到了转录组实战环节,按照惯例我会挑选10+篇比较新的带数据集的RNA-seq文章给到学徒,让大家实战。
大家需要自己去找到数据集背后的测序数据,然后使用aspera下载fq文件,走hisat2+feature流程拿到表达量矩阵,然后根据样品信息进行分组后做差异表达分析和生物学数据库注释。基本上下游分析就是我五年前的《数据挖掘》系列推文 ;
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
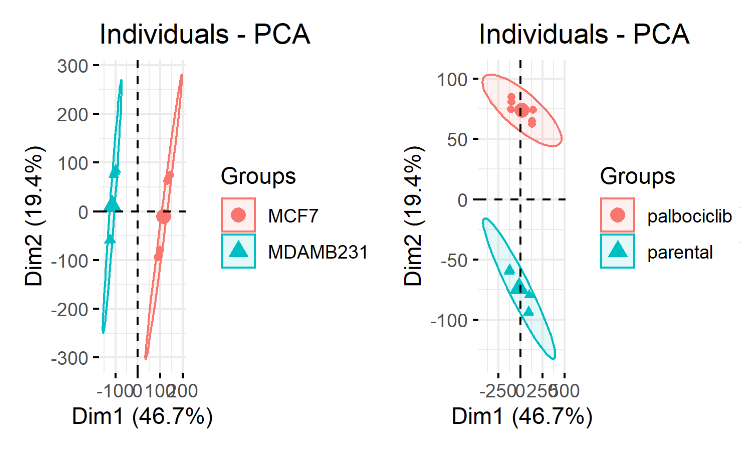
但是这次学徒完成作业后,给了我一个很诡异的PCA结果,数据集是GSE130437,如下所示:
理论上呢,这个数据集是12个样品,首先分成2个细胞系,每个细胞系都是6个样品。各个细胞系内部应该是药物组和对照组泾渭分明的。但是不知道为什么学徒做出来的第二个PCA图出现了混杂。
所以我深入检查了这12个样品的相关性矩阵热图,如下所示:
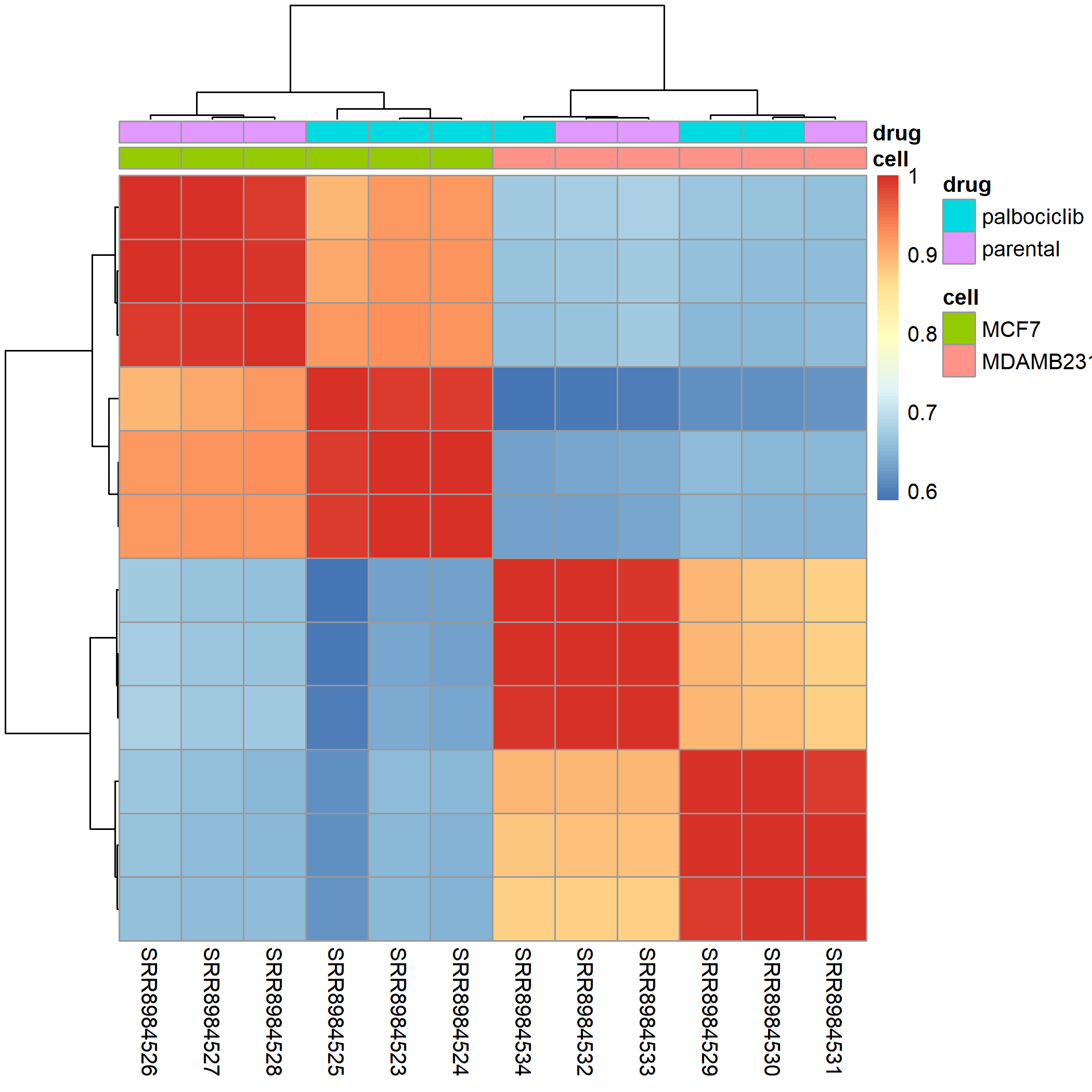
看起来呢,应该是其中一个细胞系的一个样品的分组弄错了,就是MD231细胞系里面的,而另外的那个MCF7细胞系是没有问题的哈。
所以我看到MD231细胞系差异分析结果也超级诡异,如下:
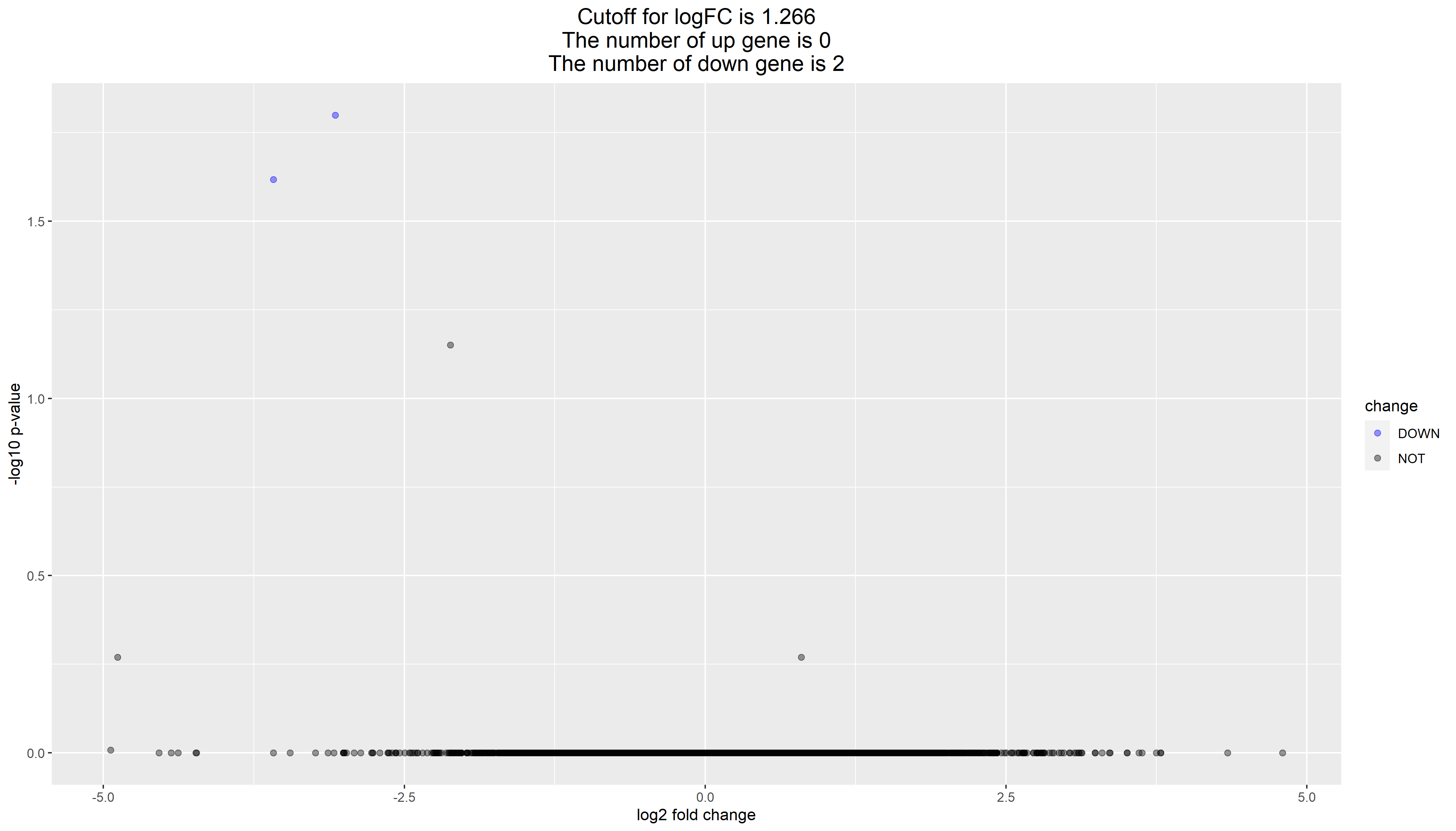
这个问题其实也很多人在公众号后台问过我,为什么他自己的差异分析拿不到任何统计学显著的基因。其实原因就是分组就搞错了,那我们继续深入检查哈。
去检查代码和数据
上游分析没有问题,就是aspera下载fq文件,走hisat2+feature流程拿到表达量矩阵,代码如下:
index=/home/jianmingzeng/reference/index/hisat/mm10/genome
hisat2=/home/jianmingzeng/biosoft/HISAT/hisat2-2.0.4/hisat2
ls raw_fq/*gz | while read id; do
$hisat2 -p 10 -x $index -U $id -S ${id%%.*}.hisat.sam
done
ls *.sam|while read id ;do (samtools sort -O bam -@ 5 -o $(basename ${id} ".sam").bam ${id});done
rm *.sam
ls *.bam |xargs -i samtools index {}
## gtf文件推荐去gencode数据库下载
gtf=/home/jianmingzeng/reference/gtf/gencode/gencode.vM12.annotation.gtf
featureCounts=/home/jianmingzeng/biosoft/featureCounts/subread-1.5.3-Linux-x86_64/bin/featureCounts
# # # 如果使用conda安装的 subread,那么featureCounts 命令应该是在环境变量的。
$featureCounts -T 5 -p -t exon -g gene_id -a $gtf -o all.id.txt *.bam 1>counts.id.log 2>&1 &
上面的代码是针对小鼠的转录组数据,如果是人类的数据,需要修改参考基因组文件索引,以及修改gencode下载的gtf文件哦。
下游分析主要是样本表型出错:
b=read.table("SraRunTable.txt",sep=",",fill = T,header=T)
b= b[,c(1,8,9,24,26)]
很有意思,确实是这个 SraRunTable.txt的样本并没有按照ID顺序排列。
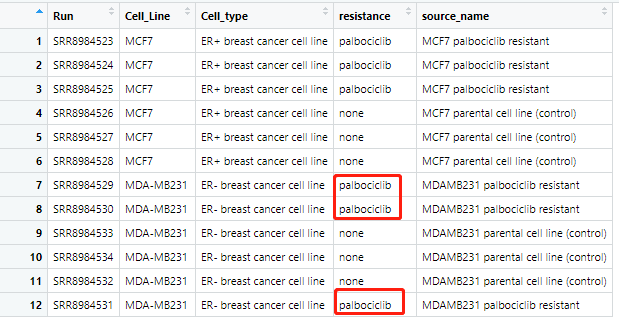
学徒的代码里面缺一个检查操作,我添加了:
identical(colnames(ensembl_matrix),b[,1])
b=b[match(colnames(ensembl_matrix),b[,1]),]
identical(colnames(ensembl_matrix),b[,1])
后面仍然是正常的分组信息啦:
group_list=b$source_name
group_list
table(group_list)
library(stringr)
phe=as.data.frame(str_split(group_list,' ', simplify = T)[,1:2])
colnames(phe)=c('cell','drug')
接下来就是我五年前的《数据挖掘》系列推文 ;
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
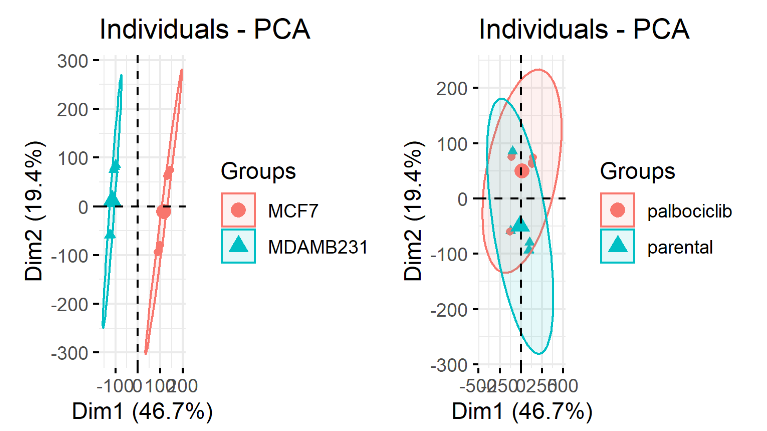
正确的结果
这样,每个细胞系后续分析都是正确的啦,从PCA上面也可以看出来。