前面我在《生信技能树》公众号分享了 对单细胞表达矩阵做gsea分析的付费代码,大受欢迎,是对指定的两个单细胞亚群进行差异分析后,根据基因的差异情况排序,然后去msigdb数据库里面进行gsea的注释,并且批量出图,在实际数据分析中非常的有用。
与此同时,不少粉丝对GSVA或者ssGSEA分析方法提出了要求,变相催稿。其实GSVA或者ssGSEA是有成熟的工具,我暂时没有找到它们的卖点。不过,我注意到了一个GitHub包,ncborcherding/escape,它提出来了对GSVA或者ssGSEA的分析结果的可视化,值得推荐。所以我们先介绍一下,假如你拿到了GSVA或者ssGSEA结果,如何可视化,这个时候呢,跟拟时序分析,转录因子分析,细胞通讯分析是大同小异的。去年我们在《生信技能树》公众号带领大家一起学习过:SCENIC转录因子分析结果的解读 ,以及:细胞通讯分析结果的解读,大家可以去读一读。
我们首先看看这个GitHub包,ncborcherding/escape文档,在:http://www.bioconductor.org/packages/release/bioc/vignettes/escape/inst/doc/vignette.html
安装方式就不再赘述,代码如下:
BiocManager::install('dittoSeq')
devtools::install_github("ncborcherding/escape")
suppressPackageStartupMessages(library(escape))
suppressPackageStartupMessages(library(dittoSeq))
suppressPackageStartupMessages(library(SingleCellExperiment))
suppressPackageStartupMessages(library(Seurat))
这个包主要是封装好了一个函数可以做gsva或者ssGSEA分析,然后对拿到的gsva或者ssGSEA分析结果进行一些可视化。
获取 MigDB中的全部基因集
MigDB中的全部基因集 都被它打包起来了,MSigDB(Molecular Signatures Database)数据库中定义了已知的基因集合:http://software.broadinstitute.org/gsea/msigdb 包括H和C1-C7八个系列(Collection),每个系列分别是:
- H: hallmark gene sets (癌症)特征基因集合,共50组,最常用;
- C1: positional gene sets 位置基因集合,根据染色体位置,共326个,用的很少;
- C2: curated gene sets:(专家)校验基因集合,基于通路、文献等:
- C3: motif gene sets:模式基因集合,主要包括microRNA和转录因子靶基因两部分
- C4: computational gene sets:计算基因集合,通过挖掘癌症相关芯片数据定义的基因集合;
- C5: GO gene sets:Gene Ontology 基因本体论,包括BP(生物学过程biological process,细胞原件cellular component和分子功能molecular function三部分)
- C6: oncogenic signatures:癌症特征基因集合,大部分来源于NCBI GEO 发表芯片数据
- C7: immunologic signatures: 免疫相关基因集合。
GS <- getGeneSets(library = "H") GSMigDB中的全部基因集 被 构建成为: a list of GSEABase GeneSet objects ,获取 hallmark gene sets (癌症)特征基因集合,共50组的代码如上所示:
- http://www.bio-info-trainee.com/2105.html
- http://www.bio-info-trainee.com/2102.html
- https://www.gsea-msigdb.org/gsea/msigdb
- https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp
运行通用函数:
enrichIt()做gsva或者ssGSEA分析sce # 可以是SingleCellExperiment or Seurat # 或者一个简单的矩阵 ES <- enrichIt(obj = sce, gene.sets = GS, groups = 1000, cores = 2) # 返回了 ES 这个矩阵作者在这里包装了一个通用函数:
enrichIt()can handle either a matrix of raw count data or will pull that data directly from a SingleCellExperiment or Seurat object.GSVA或者ssGSEA结果本质上也是一个矩阵
在运行gsva或者ssGSEA分析之前,我们的矩阵是单细胞表达量矩阵,2万多个基因在成百上千个细胞里面的表达量矩阵,主要是走质控降维聚类分群和细胞亚群的生物学注释流程,可以看看我们前面的例子:人人都能学会的单细胞聚类分群注释 。
但是做完了GSVA或者ssGSEA分析之后呢,结果本质上也是一个矩阵,这个时候这个矩阵并不需要走质控降维聚类分群和细胞亚群的生物学注释流程。但是可以借鉴我五年前写的表达矩阵的15种可视化方式, 非常丰富:http://bio-info-trainee.com/tmp/basic_visualization_for_expression_matrix.html可视化之1热图
封装了一个 dittoHeatmap 函数,效果如下所示:
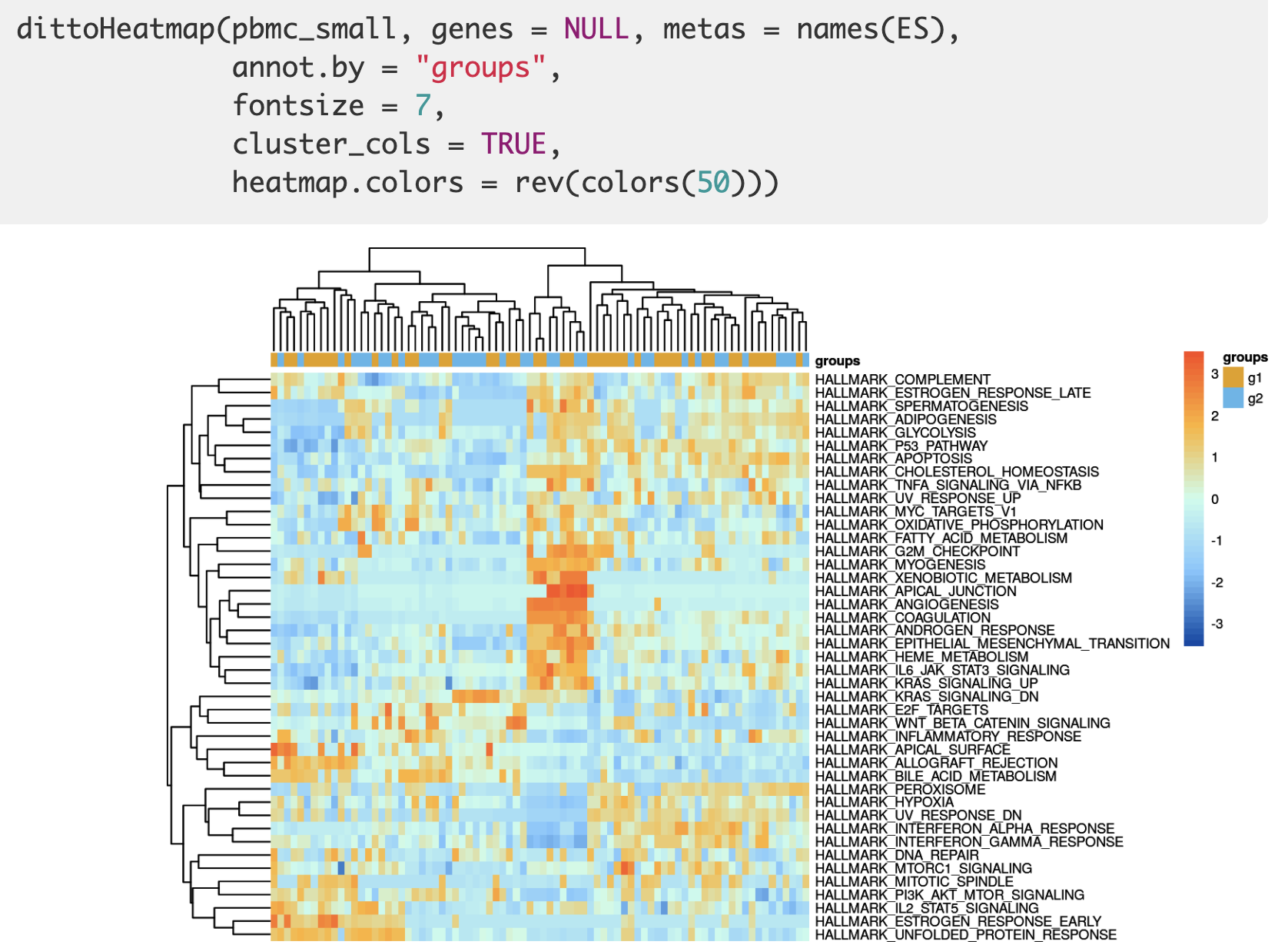
当然了,如果是觉得hallmark gene sets (癌症)特征基因集合,共50组,数量太多了,也可以挑选自己认识的少量几个通路集中可视化。可视化之2 箱线图或者小提琴图
封装了一个 multi_dittoPlot函数,效果如下所示:
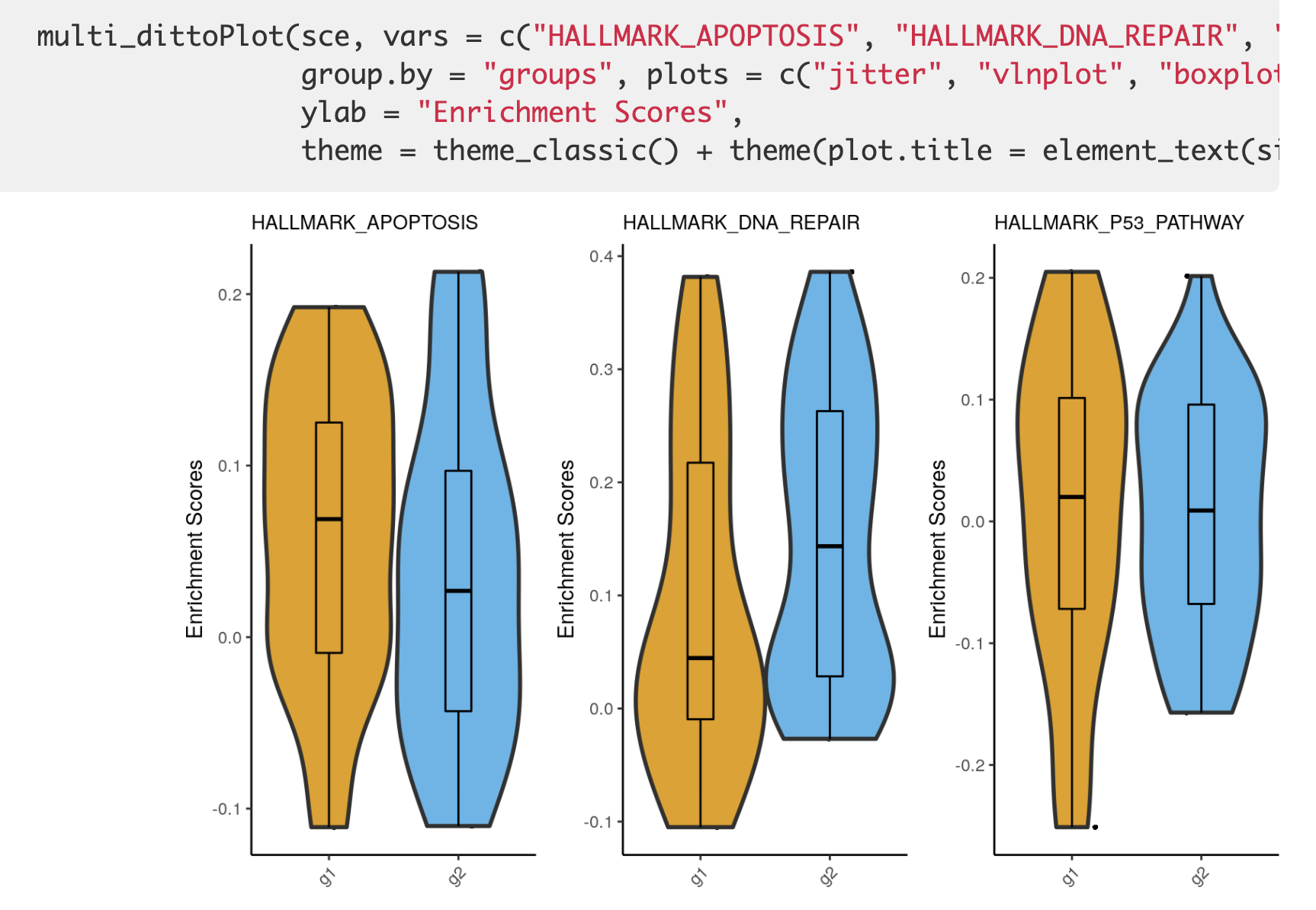
这个时候呢,因为信息密度不如热图,所以通常是并不建议可视化全部的通路,抓住重点即可,而且呢,这个时候最好是有一个分组信息,说清楚自己的目标,就是想看两个不同的细胞亚群的某一些生物学功能的GSVA或者ssGSEA结果差异。可视化之3 热力图
如果你认为两个生物学通路会有规律,就可以做一下这个热力图。
封装了一个 dittoScatterHex 函数,效果如下所示:
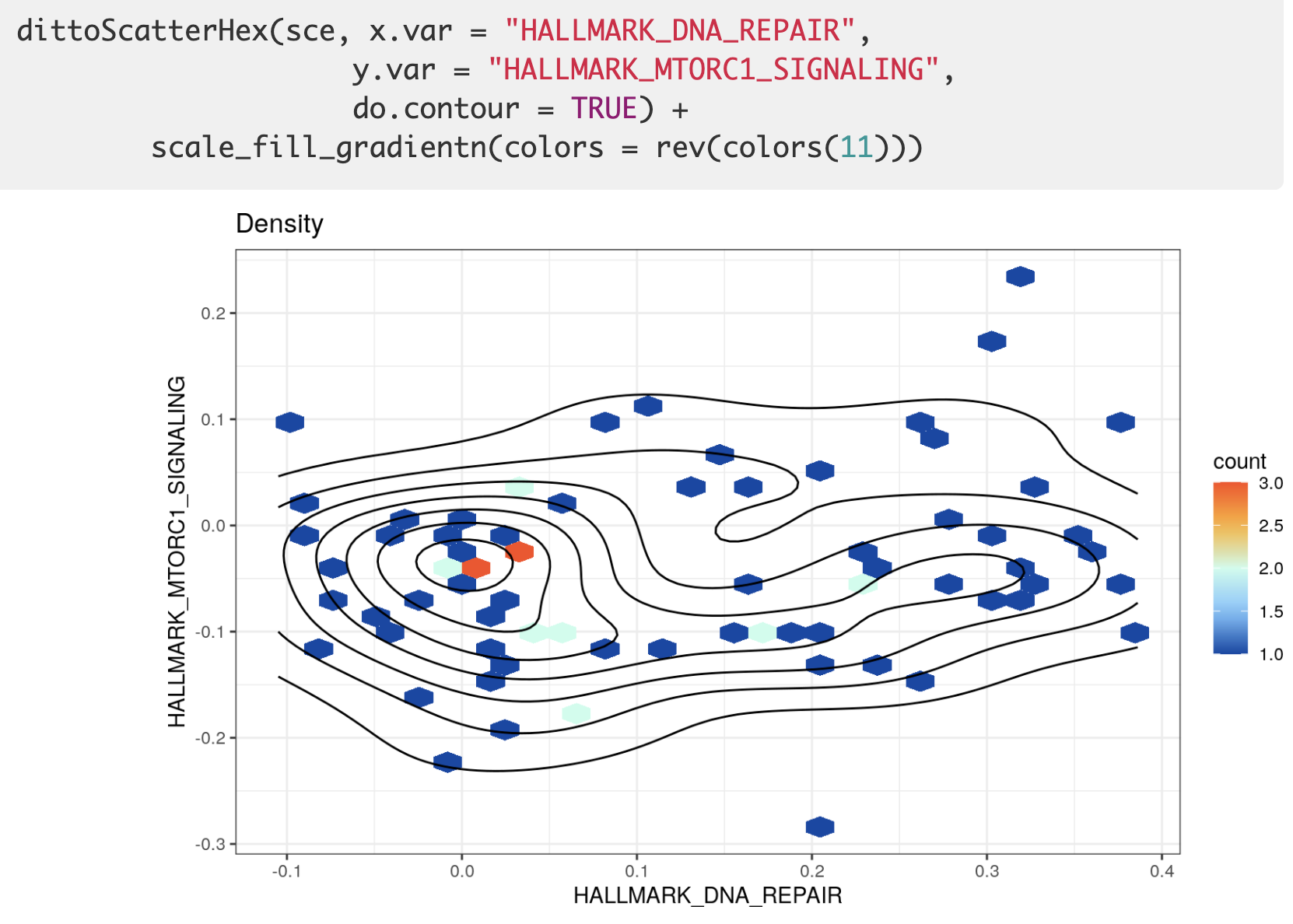
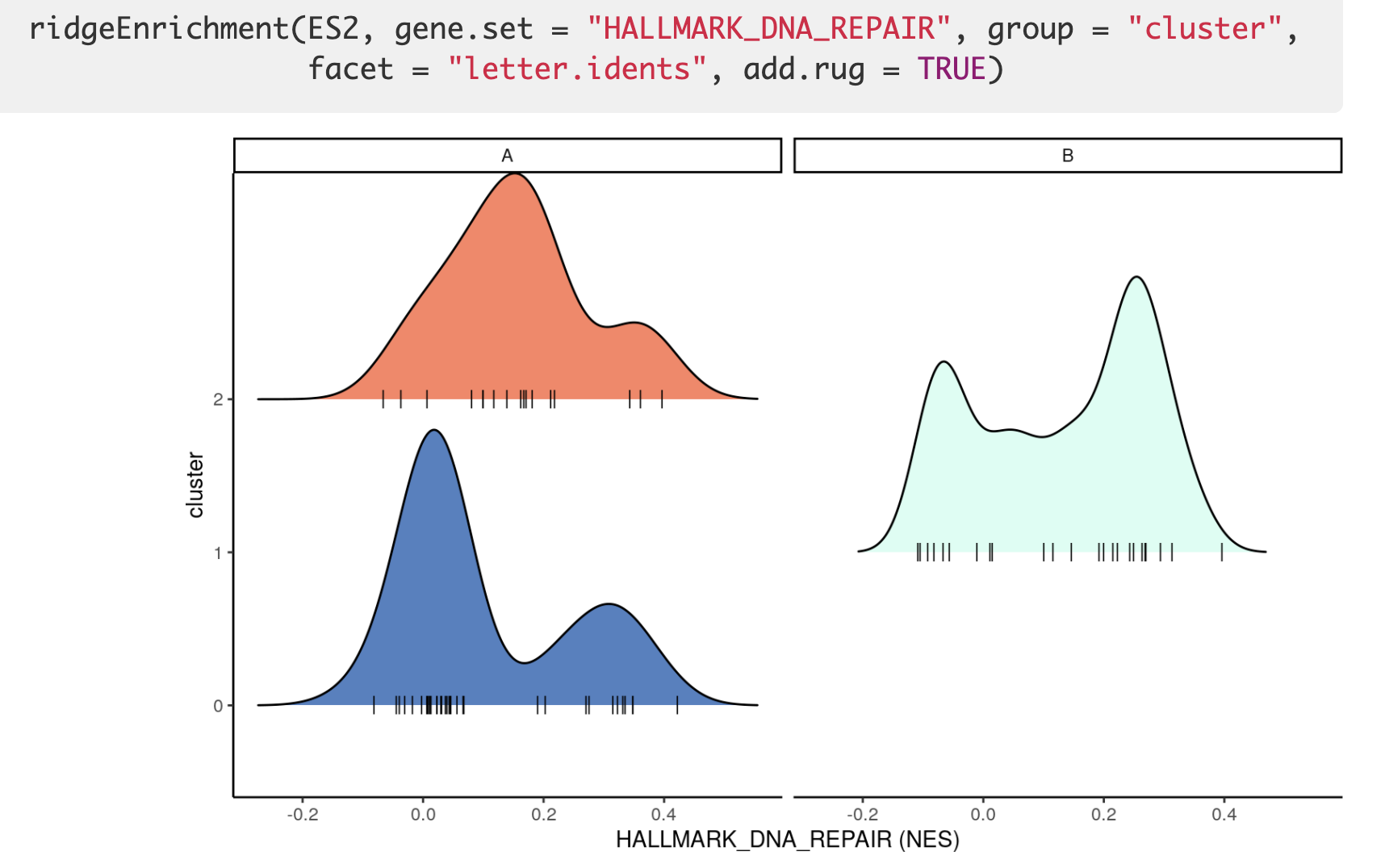
可视化之4地毯图/山峰图
封装了一个 ridgeEnrichment 函数,效果如下所示:
可视化之5主成分方法图
这个时候首先要把GSVA或者ssGSEA结果矩阵进行PCA分析,然后才能继续可视化,代码如下:
PCA <- performPCA(enriched = ES2, groups = c("groups", "cluster")) pcaEnrichment(PCA, PCx = "PC1", PCy = "PC2", contours = TRUE) pcaEnrichment(PCA, PCx = "PC1", PCy = "PC2", contours = FALSE, facet = "cluster") masterPCAPlot(ES2, PCx = "PC1", PCy = "PC2", top.contribution = 10)这个时候有3种可视化,最后那个masterPCAPlot函数的可视化效果如下所示:
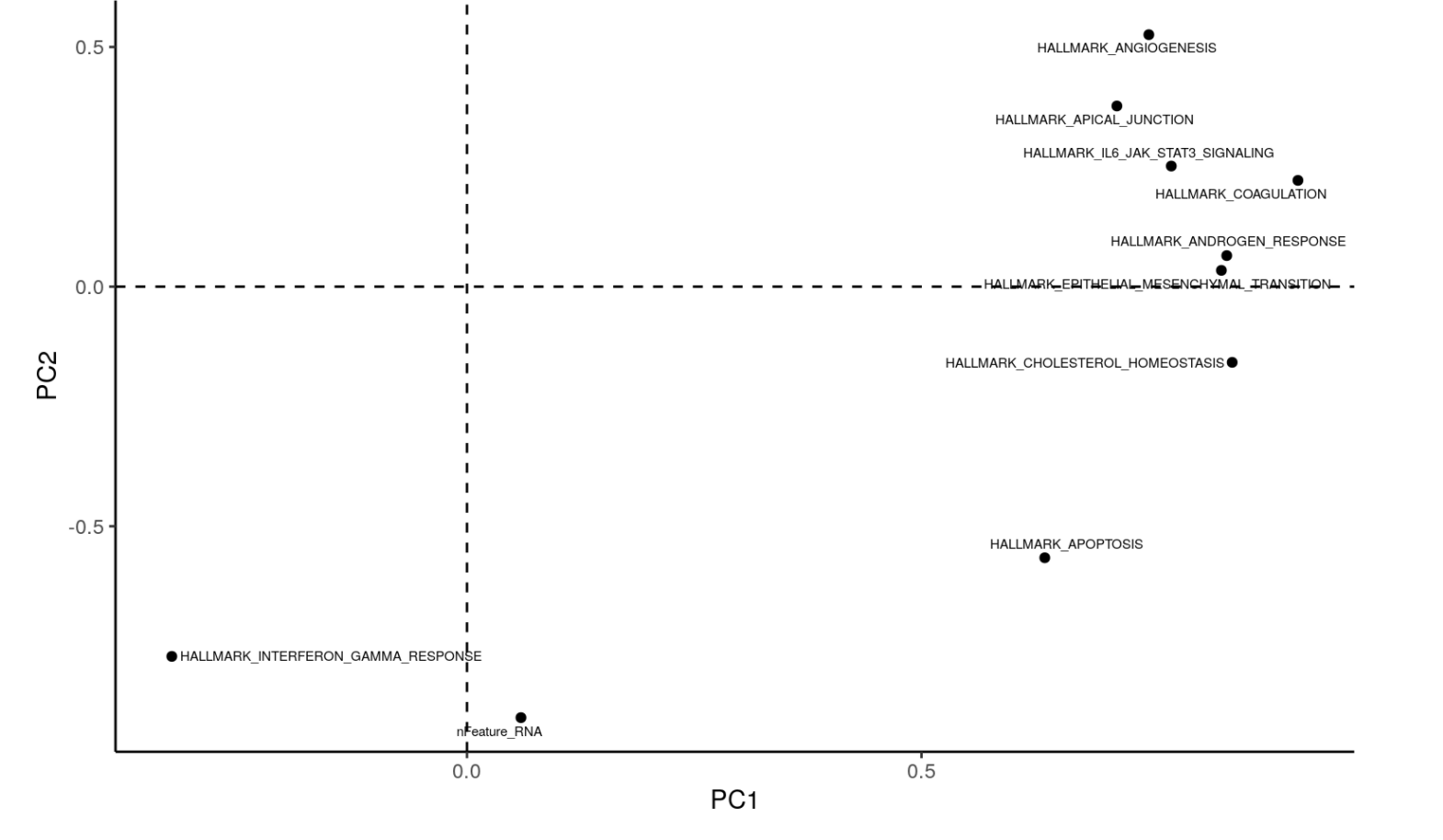
实际上,就是最开始的热图上面的行的层次聚类的另外一种表现形式啦。统计分析之差异
封装了一个 getSignificance 函数,比如可以看看前面hallmark gene sets (癌症)特征基因集合,共50组的GSVA或者ssGSEA结果矩阵,在我们已经定义好的细胞亚群的差异。
这样的可视化R包难度有多大呢?
去年我们在《生信技能树》公众号带领大家一起学习过:SCENIC转录因子分析结果的解读 ,以及:细胞通讯分析结果的解读,大家可以去读一读。其实它们最后也会输出一个结果矩阵,跟GSVA或者ssGSEA结果矩阵并没有本质上的区别,从可视化角度来看,反而最大的门槛在于生物学解释。
开发这样的可视化R包难度其实并不大,因为原创性并不多,一个包想流行,大家觉得靠的是什么呢?