前面我们分享了 跟着Nature Medicine学MeDIP-seq数据分析,数据和代码都是公开,这个2G的压缩包文件,足以学习3个月,写60篇教程。
这里面的MeDIP-seq指的是DNA,那么MeRIP-seq其实就是RNA水平的又叫做m6a测序,恰好看到了咱们的表观微信交流群我们的生信技能树优秀转录组讲师在分享全套MeRIP-seq文章图表复现代码,我借花献佛整理一下分享给大家:
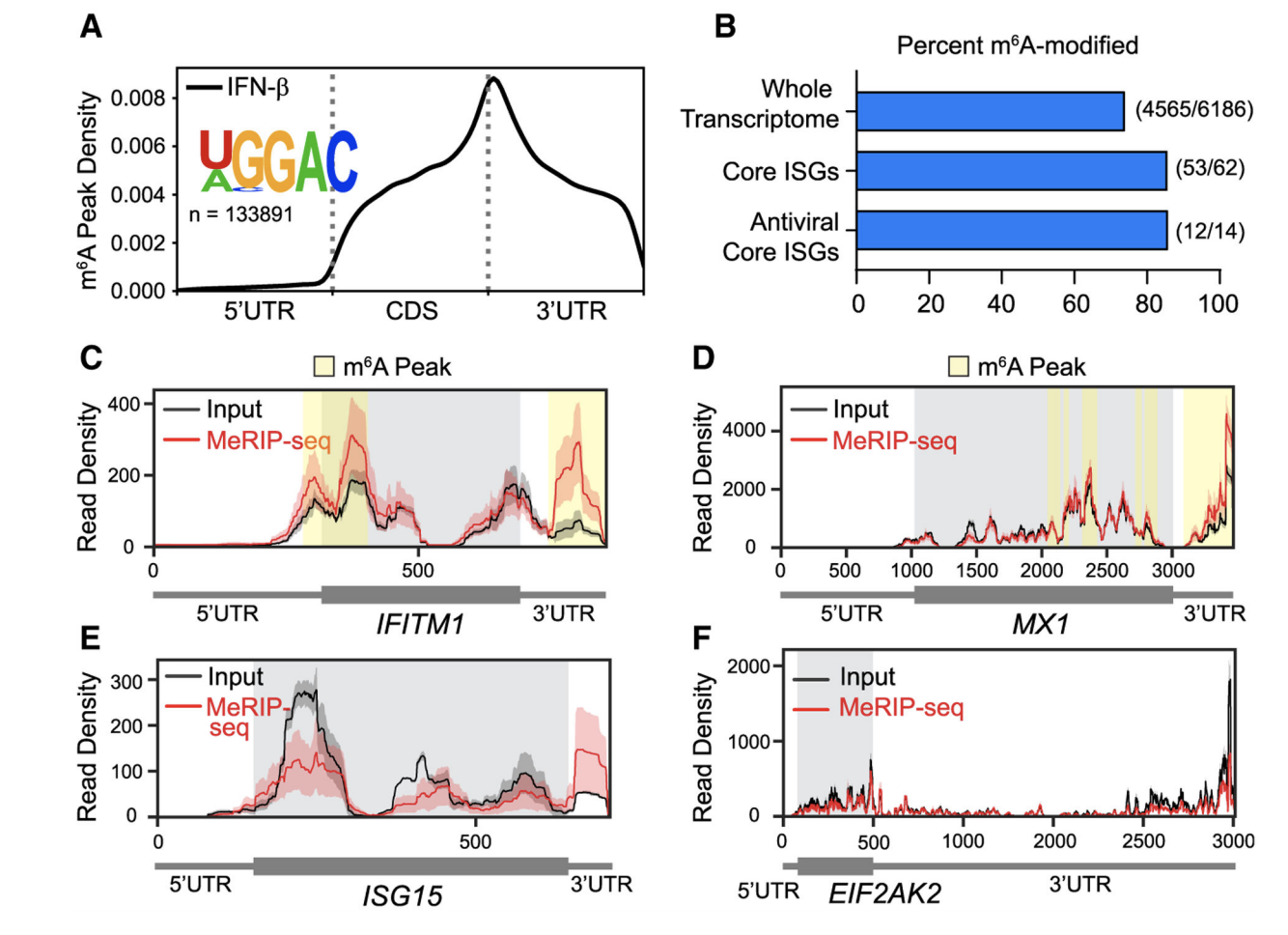
这个代码关联到了两个 文章,首先是Cell Rep. 2021 Mar ,标题是:《Post-transcriptional regulation of antiviral gene expression by N6-methyladenosine》,然后是文章《Limits in the detection of m6A changes using MeRIP/m6A-seq》
数据在:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE155413 , 是12个样品:
GSM4704026 Ifn input rep1
GSM4704027 Ifn input rep2
GSM4704028 Ifn input rep3
GSM4704029 Ifn meRIP rep1
GSM4704030 Ifn meRIP rep2
GSM4704031 Ifn meRIP rep3
GSM4704032 Mock input rep1
GSM4704033 Mock input rep2
GSM4704034 Mock input rep3
GSM4704035 Mock meRIP rep1
GSM4704036 Mock meRIP rep2
GSM4704037 Mock meRIP rep3
这个实验设计是超级严谨了,其实就Ifn处理前后的两个组,但是每个组都有3个重复,而且是非常严格的MeRIP和 input方便找peaks。
最重要的是,提供了全套MeRIP-seq 图表复现代码在GitHub: https://github.com/al-mcintyre/merip_reanalysis_scripts 这个也是接近2G的压缩包!
首先是文章的重要的图表绘制的R代码 :
2.7K Jan 9 2020 plot_fig1b.R
7.6K Jan 9 2020 plot_fig2.R
20K Jan 9 2020 plot_fig3.R
4.0K Jan 9 2020 plot_fig3_helper.R
11K Jan 9 2020 plot_fig4.R
4.7K Jan 9 2020 plot_fig4_mergedpeaks.R
3.6K Jan 9 2020 plot_fig5.R
6.4K Jan 9 2020 plot_sfig1a.R
4.4K Jan 9 2020 plot_sfig1g.R
1.0K Jan 9 2020 plot_sfig2.R
368B Jan 9 2020 rhelper.R
9.9K Jan 9 2020 run_metdiff_peakcaller.R
然后是具体数据分析的Linux上游的shell脚本:
830B Jan 9 2020 merged_peak_analysis.sh
1.5K Jan 9 2020 plot_fig1a.sh
1.3K Jan 9 2020 plot_fig1bc.sh
3.8K Jan 9 2020 plot_fig2.sh
926B Jan 9 2020 plot_fig4prep.sh
931B Jan 9 2020 plot_sfig2.sh
7.9K Jan 9 2020 run_full_analysis.sh
1.3K Jan 9 2020 run_kallisto.sh
921B Jan 9 2020 run_macs2.2.sh
3.1K Jan 9 2020 run_star.1.sh
研究者自己擅长python,所以也有一些配套的python小脚本:
4.4K Jan 9 2020 get_most_highly_expressed.py
3.4K Jan 9 2020 plot_fig1a_counts_vs_expression.py
799B Jan 9 2020 plot_fig1a_summary.py
1.4K Jan 9 2020 plot_fig1c_replicate_overlap.py
图的颜值不错:
很早以前我就在《生信技能树》发布过教程:新的ngs流程该如何学习(以CUT&Tag 数据处理为例子),提到了我自己是不太可能去把所有的ngs流程全部录制视频的,只能说是更好的传达学习方法给到大家。其实如果你看过我表观组学,比如《ChIP-seq数据分析》 和 《ATAC-seq数据分析》 就会发现其实这个m6A数据处理大同小异的,当然了,肯定是会有一些细微差异是需要注意的。具体数据分析的Linux上游的shell脚本看研究者的GitHub :
7.9K Jan 9 2020 run_full_analysis.sh
1.3K Jan 9 2020 run_kallisto.sh
921B Jan 9 2020 run_macs2.2.sh
3.1K Jan 9 2020 run_star.1.sh
可以看到首先是 trimmomatic 这个java软件进行fastq文件的质量控制:
java -jar trimmomatic-0.36.jar PE -phred33 $PRETRIM1 $PRETRIM2 $FASTQ1 fastqs/${f}_R1_unpaired.fq.gz $FASTQ2 fastqs/${f}_R2_unpaired.fq.gz ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:15
然后是STAR这个软件进行比对:
STAR --genomeDir $STAR_INDEX --runThreadN $NUM_THREADS --readFilesIn fastqs/${f}_R1.fastq.gz fastqs/${f}_R2.fastq.gz --outFilterMultimapNmax 1 --readFilesCommand zcat --outFileNamePrefix ${f}.star. --genomeLoad LoadAndKeep --outSAMstrandField intronMotif
还有kallisto这个软件进行定量
kallisto quant -t $NUM_THREADS -i $IDX -o kallisto_results/$SAMPLE.kallisto fastqs/${COND}_input_${REP}_R1.trim.fastq.gz fastqs/${COND}_input_${REP}_R2.trim.fastq.gz
到目前为止,其实都是跟转录组流程类似,但是因为有 macs2软件进行 callpeak 代码是:
macs2 callpeak -t alignments/${COND}_IP_${REP}.star.sorted.bam -c alignments/${COND}_input_${REP}.star.sorted.bam --nomodel --extsize $FRAGLEN -g 100e6 -n macs2_results/${COND}_${REP}.macs2 -f BAM --verbose 3
这一切代码能成功运行的前提是Linux基础,再怎么强调生物信息学数据分析学习过程的计算机基础知识的打磨都不为过,我把它粗略的分成基于R语言的统计可视化,以及基于Linux的NGS数据处理:
把R的知识点路线图搞定,如下:
- 了解常量和变量概念
- 加减乘除等运算(计算器)
- 多种数据类型(数值,字符,逻辑,因子)
- 多种数据结构(向量,矩阵,数组,数据框,列表)
- 文件读取和写出
- 简单统计可视化
- 无限量函数学习
Linux的6个阶段也跨越过去 ,一般来说,每个阶段都需要至少一天以上的学习:
- 第1阶段:把linux系统玩得跟Windows或者MacOS那样的桌面操作系统一样顺畅,主要目的就是去可视化,熟悉黑白命令行界面,可以仅仅以键盘交互模式完成常规文件夹及文件管理工作。
- 第2阶段:做到文本文件的表格化处理,类似于以键盘交互模式完成Excel表格的排序、计数、筛选、去冗余,查找,切割,替换,合并,补齐,熟练掌握awk,sed,grep这文本处理的三驾马车。
- 第3阶段:元字符,通配符及shell中的各种扩展,从此linux操作不再神秘!
- 第4阶段:高级目录管理:软硬链接,绝对路径和相对路径,环境变量。
- 第5阶段:任务提交及批处理,脚本编写解放你的双手。
- 第6阶段:软件安装及conda管理,让linux系统实用性放飞自我。