拿多个病人的多个样本进行单细胞转录组测序是非常正常的,比如下面的:
- 发表在 Nat Med. 2018 Aug; 题目是:Phenotype molding of stromal cells in the lung tumor microenvironment. 共选取5例病人的共19个样本,通过10×genomics单细胞转录组测序探索基质细胞的亚群分类、基因功能(信号通路)、关键marker基因和临床预后,共鉴定出52个基质细胞亚群,
- 发表在 Nature Medicine (2018) ,标题是:Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis 作者从3个乳腺癌患者体内通过FACS筛选到乳腺癌中肿瘤浸润淋巴细胞(TILs),使用商业仪器10X来做单细胞转录组,得到6,311个T细胞数据。
- 还有我们在单细胞天地分享的scRNA-seq揭示胰腺导管腺癌的瘤内异质性和恶性进展 ,有24个原发性PDAC肿瘤病人样本及11个对照胰腺样本(3例非胰腺肿瘤患者和8例非恶性胰腺肿瘤患者样本)
这些2018年的研究,大多数都是百万经费起的项目,所以基本上是CNS级别的成果。 现在想发普通的单细胞文章,也是得做多个样本了,就面临如何整合的问题,其中最出名的当然是Seurat包的CCA方法了,具体多火呢,发了才一年,引用就快破千!
这样的单细胞转录组测序整合算法超级多,我想你应该是不会一一细看的,我就列出来而已:
- MNNcorrect (https://doi.org/10.1038/nbt.4091)
- CCA + anchors (Seurat v3) (https://doi.org/10.1101/460147)
- CCA + dynamic time warping (Seurat v2) (https://doi.org/10.1038/nbt.4096)
- LIGER (https://doi.org/10.1101/459891)
- Harmony (https://doi.org/10.1101/461954)
- Conos(https://doi.org/10.1101/460246)
- Scanorama(https://doi.org/10.1101/371179)
- scMerge(https://doi.org/10.1073/pnas.1820006116)
今天就从简单的Seurat包的merge函数到,CCA + anchors (Seurat v3),再到Harmony 给大家介绍一下哈。
因为一直听说harmony效果更佳,今天才有空所以想体验一下,比如 2019的 Cell的文章《Landscap and Dynamics of Single Immune Cells in Hepatocellular Carcinoma》, 整合两类数据(包括SMART-seq2和10X),就使用的是 harmony算法。
结果在“百度百科”查到它:据英国《每日邮报》2017年4月4日报道,RealDoll公司设计出世界上首款性爱机器人,可以和用户产生情感交流。Harmony 2.0配有苏格兰口音,即将上市,售价预计为1万美元。(如果有关于Harmony的用户体验欢迎交流哈!)
先看看简单的Seurat包的merge函数
首先保证你的 folder 变量存储着全部的10x单细胞转录组数据的cellranger结果哈;
# folder 这个变量,存储着多个10x单细胞转录组数据的cellranger结果
library(Seurat)
sceList = lapply(folders,function(folder){
CreateSeuratObject(counts = Read10X(folder),
project = folder )
})
sce.big <- merge(sceList[[1]],
y = c(sceList[[2]],sceList[[3]],sceList[[4]],sceList[[5]],
sceList[[6]],sceList[[7]],sceList[[8]]),
add.cell.ids = folders,
project = "mouse8")
sce.big
table(sce.big$orig.ident)
save(sce.big,file = 'sce.big.merge.mouse8.Rdata')
然后Seurat包的CCA整合
# folder 这个变量,存储着多个10x单细胞转录组数据的cellranger结果
library(Seurat)
sceList = lapply(folders,function(folder){
CreateSeuratObject(counts = Read10X(folder),
project = folder )
})
sceList
# 标注化和寻找 VariableFeatures
for (i in 1:length(sceList)) {
print( Sys.time() )
sceList[[i]] <- NormalizeData(sceList[[i]], verbose = FALSE)
sceList[[i]] <- FindVariableFeatures(sceList[[i]], selection.method = "vst",
nfeatures = 2000, verbose = FALSE)
}
sceList
Sys.time()
# 整合 二个样本
sce.anchors <- FindIntegrationAnchors(object.list = sceList, dims = 1:30)
# load('sce.anchors.RDATA')
Sys.time()
sce.integrated <- IntegrateData(anchorset = sce.anchors, dims = 1:30)
Sys.time()
library(ggplot2)
library(cowplot)
# switch to integrated assay. The variable features of this assay are automatically
# set during IntegrateData
DefaultAssay(sce.integrated) <- "integrated"
# 接下来的分析,都基于 sce.integrated 啦
接着是MNNs算法
Seurat主要是处理10x单细胞转录组数据,而10x仪器商业上的成功可以说是成就了Seurat包,另外一个比较火的多个样本单细胞转录组数据整合算法是mutual nearest neighbors (MNNs)
因为并不是必须,所以我一直没有使用它!单细胞转录组数据的整合分析才是主流,所以我在生信技能树的多个单细胞转录组样本的数据整合之CCA-Seurat包 是非常值得大家细看的,而且使用scran包的MNN算法来去除多个单细胞转录组数据批次效应 也展现了一个很好的例子。
最后是我们今天的主角harmony
发表这个harmony算法的文章是2019年11月的《Fast, sensitive and accurate integration of single-cell data with Harmony》,目前仍然是一个GitHub包,在 https://github.com/immunogenomics/harmony 所以安装它的方式是:
library(devtools)
install_github("immunogenomics/harmony")
前面的Seurat标准流程仍然是一样的:
sce <- NormalizeData(sce, normalization.method = "LogNormalize",
scale.factor = 1e4)
GetAssay(sce,assay = "RNA")
sce <- FindVariableFeatures(sce,
selection.method = "vst", nfeatures = 2000)
sce <- ScaleData(sce)
sce <- RunPCA(object = sce, pc.genes = VariableFeatures(sce))
DimHeatmap(sce, dims = 1:12, cells = 100, balanced = TRUE)
ElbowPlot(sce)
这个时候的sce对象,里面存储这多个10x样品的单细胞结果,以 orig.ident 列作为区分。
接下来开始引入harmony步骤,超级简单:
library(harmony)
seuratObj <- RunHarmony(sce, "orig.ident")
names(seuratObj@reductions)
seuratObj <- RunUMAP(seuratObj, dims = 1:15,
reduction = "harmony")
DimPlot(seuratObj,reduction = "umap",label=T )
对harmony整合好的对象进行聚类分群:
sce=seuratObj
sce <- FindNeighbors(sce, reduction = "harmony",dims = 1:15)
sce <- FindClusters(sce, resolution = 0.1)
table(sce@meta.data$seurat_clusters)
DimPlot(sce,reduction = "umap",label=T)
DimPlot(sce,reduction = "umap",label=T,
group.by = 'orig.ident')
可以看到如下所示的结果,上图是整合前,下图整合后 :
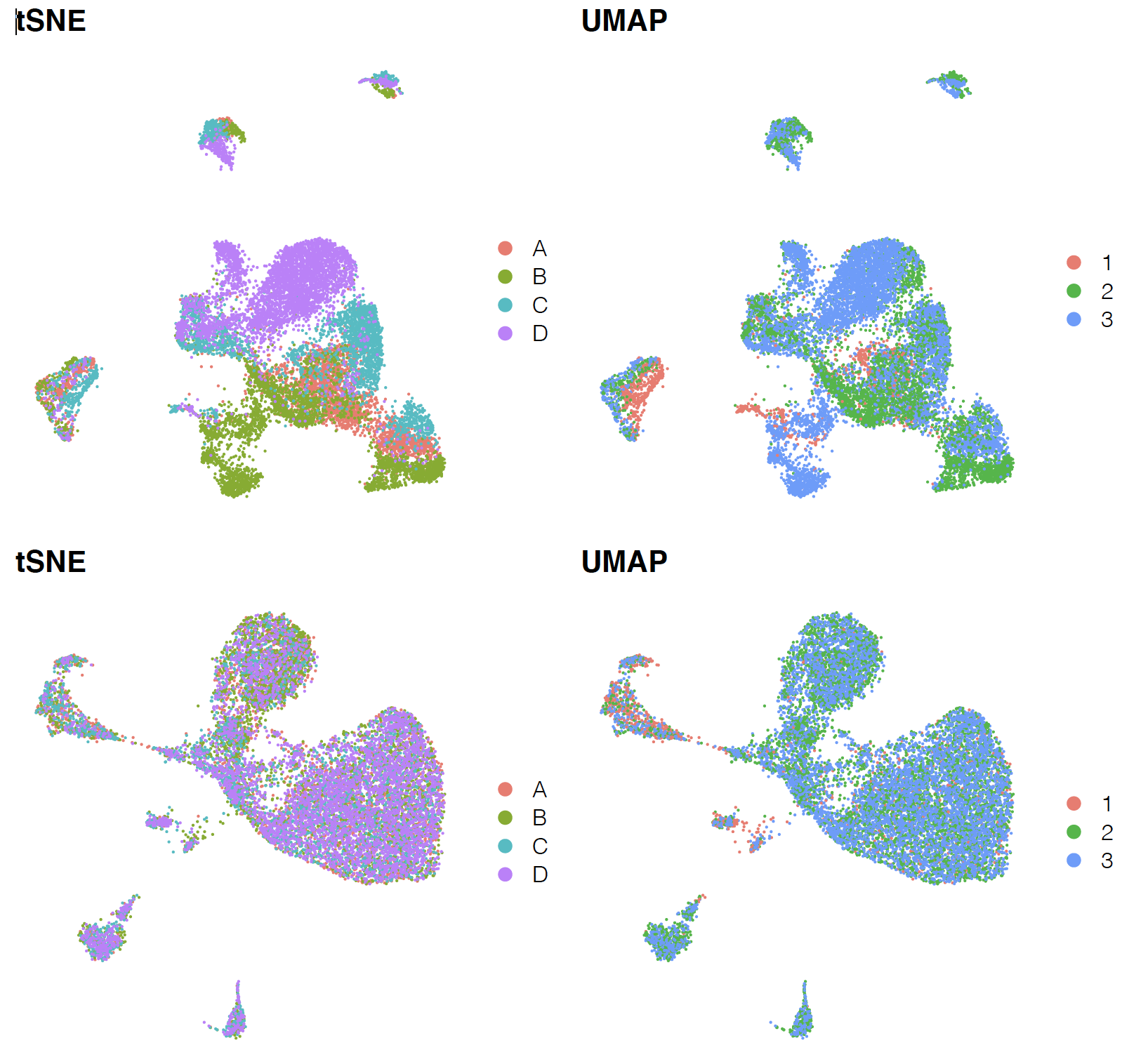
如果你感兴趣这个harmony技术的细节,建议看前两天在《单细胞天地》的 《单细胞分析十八般武艺1:harmony》
类似的容易让人想歪的生物信息学词条好像还不少
比如: julia 这个编程语言,如果你 直接搜索它!

哈哈,群体的智慧是无穷的,欢迎各路神通广大的朋友留言给出类似的例子!