RNA-seq数据分析绝大部分小伙伴应该都是问题不大了,我在B站也有教学视频,视频观看方式 :
-
视频免费在B站:https://www.bilibili.com/video/BV12s41137HY 大家学习的时候记得发弹幕交流哈。
-
也有微云离线版本视频下载本地播放:
-
- 上游分析视频以及代码资料在:https://share.weiyun.com/5QwKGxi
- 下游主要是基于counts矩阵的标准分析的代码 https://share.weiyun.com/50hfuLi
-
RNA-SEQ实战演练的素材:https://share.weiyun.com/5h1Z2QY ,包括一些公司PPT,综述以及文献以及测试数据
-
RNA-SEQ 实战演练的思维导图:文档链接:https://mubu.com/doc/38y7pmgzLg 密码:p6fo
转录组的标准分析,比较容易复现,基本上看我六年前的表达芯片的公共数据库挖掘系列推文即可;
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
但是这些年转录组并不是毫无进步,其中一个最大的转变就是进阶成为了单细胞转录组。另外一个值得一提的就是各路新工具层出不穷,比如使用Glimma 交互式可视化RNA-seq数据。主要是在在两个关键时刻,让你的可视化灵动起来,其bioconductor官方链接是:https://bioconductor.org/packages/release/bioc/html/Glimma.html
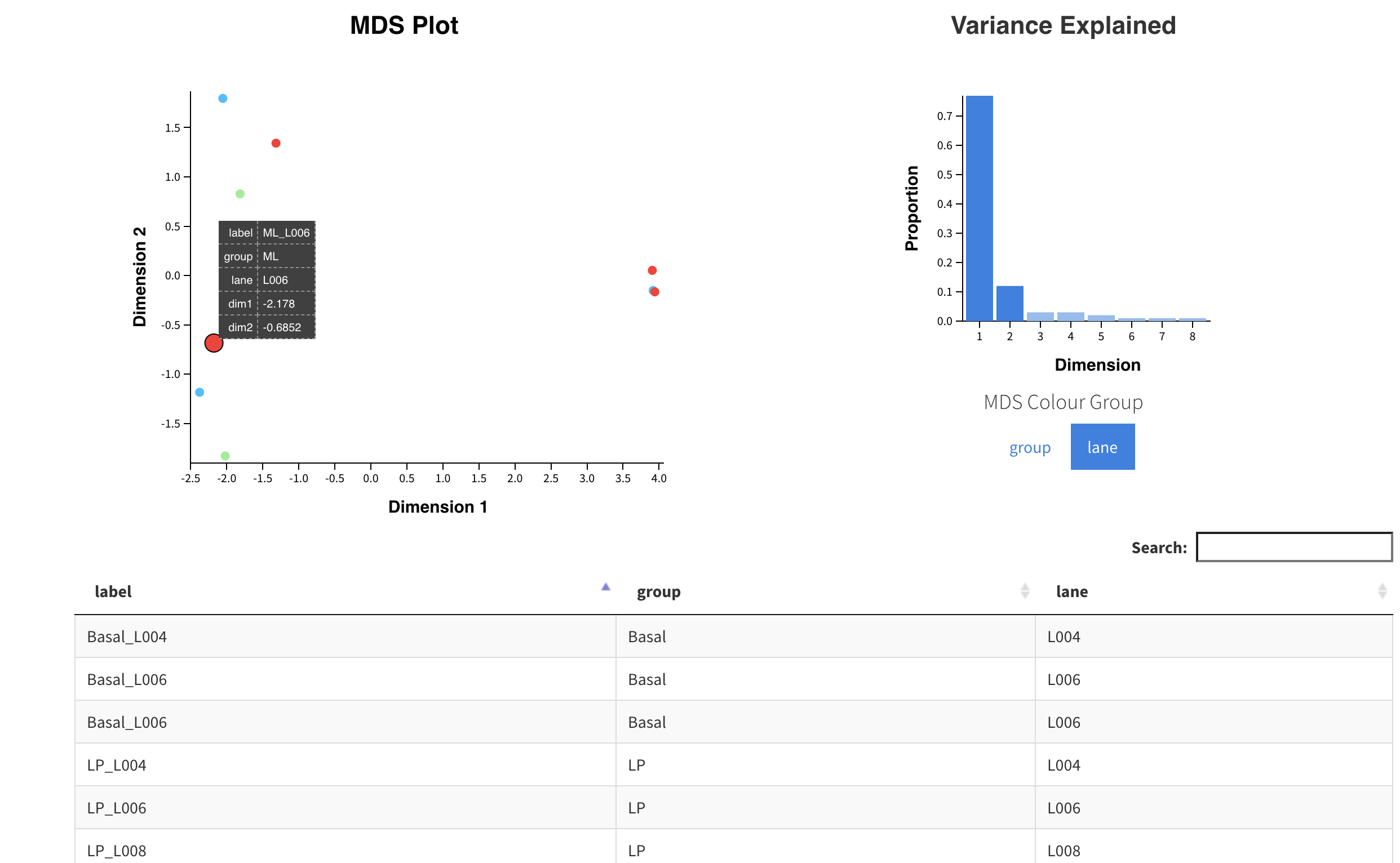
PCA或者MDS看组间差异和组内差异
虽然这个Glimma 交互式可视化RNA-seq数据采用的是MDS的方法,不过本质上跟PCA并没有太多区别,主要都是在二维平面上看看组间差异和组内差异是否符合实验设计。
通常呢,同一个分组的多个样品在这个二维画布上面是需要尽可能的靠拢,而不同组需要尽可能的远离。如下所示是一个比较好的例子:
MD或者火山图看差异基因分布情况
火山图大家应该是也基本上都没有问题,下面的MA图其实跟火山图非常的类似,两者都是log2FC信息,不同的是火山图展现P值,而MA图展现的是表达量情况
- 火山图是为了说明log2FC比较大的一般来说具有统计学显著性
- 而MA图是为了说明log2FC无论大小,都不应该与表达量有相关性。
我们通常呢,挑选差异基因,会选择那些log2FC比较大而且具有统计学显著性的上下调基因,不过加上MA图,就可以进一步挑选那些表达量也比较高的,因为这样的基因呢,容易去实验验证。而且呢,通常情况下常识会告诉我们高表达量基因更容易发挥作用。
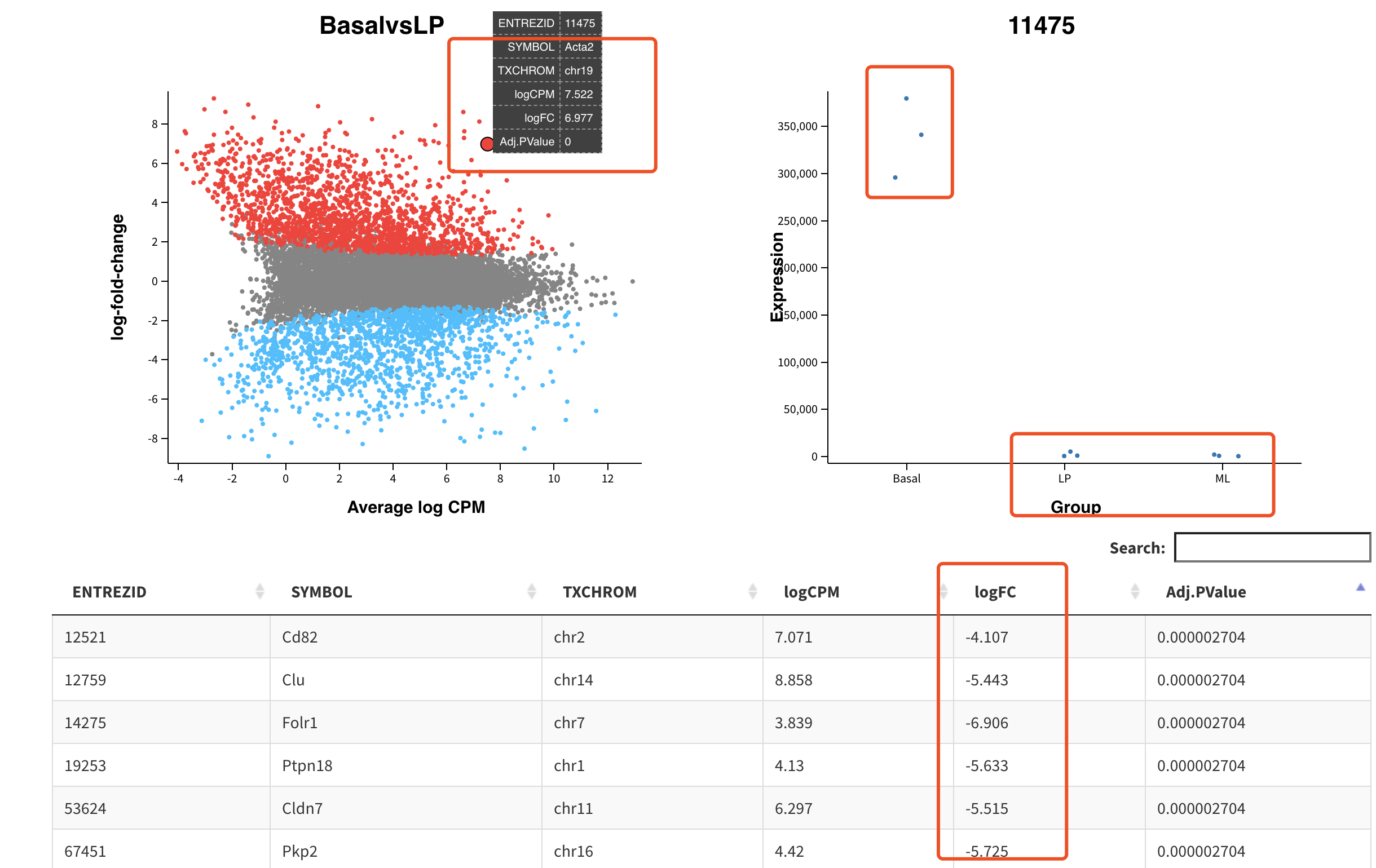
这个Glimma 交互式可视化RNA-seq数据优势在于,它不仅仅是给出数值,而且是可以交互式的具体看某个基因是如何的差异!
如下所示的测试代码:
library(limma)
y <- matrix(rnorm(10000*9),10000,9)
fivenum(y)
(group=rep(LETTERS[1:3],each=3) )
y[sample(1:10000,1000),1:3]=y[sample(1:10000,1000),1:3]+2
y[sample(1:10000,1000),4:6]=y[sample(1:10000,1000),4:6]+2
y[sample(1:10000,1000),7:9]=y[sample(1:10000,1000),7:9]+2
library(pheatmap)
# pheatmap(y )
design <- model.matrix(~0+group)
colnames(design) <- gsub("group", "", colnames(design))
design
contr.matrix <- makeContrasts(
AVSB = A-B,
AVSC = A-C,
BVSC = B-C,
levels = colnames(design))
contr.matrix
vfit <- lmFit(y, design)
vfit <- contrasts.fit(vfit, contrasts=contr.matrix)
efit <- eBayes(vfit)
plotSA(efit)
colnames(efit)
summary(decideTests(efit))
AVSB <- topTreat(efit, coef=1, n=Inf)
head(AVSB)
y=as.data.frame(y)
genes=data.frame(id=rownames(y))
head(genes)
head(y)
library(Glimma)
plotMD(tfit, column=1, status=dt[,1], main=colnames(tfit)[1],
xlim=c(-8,13))
glMDPlot(tfit, coef=1, status=dt[,1],
main=colnames(tfit)[1], counts= y ,
samples= colnames(y),
anno=genes,
groups=group,
#id.column="ENTREZID",
#display.columns=c("SYMBOL", "ENTREZID"),
#search.by="SYMBOL",
launch=FALSE)
这个 glMDPlot 函数运行成功后,会在你的R工作目录输出一个文件夹哦,里面就有一个网页可以使用你的浏览器打开即可查看:
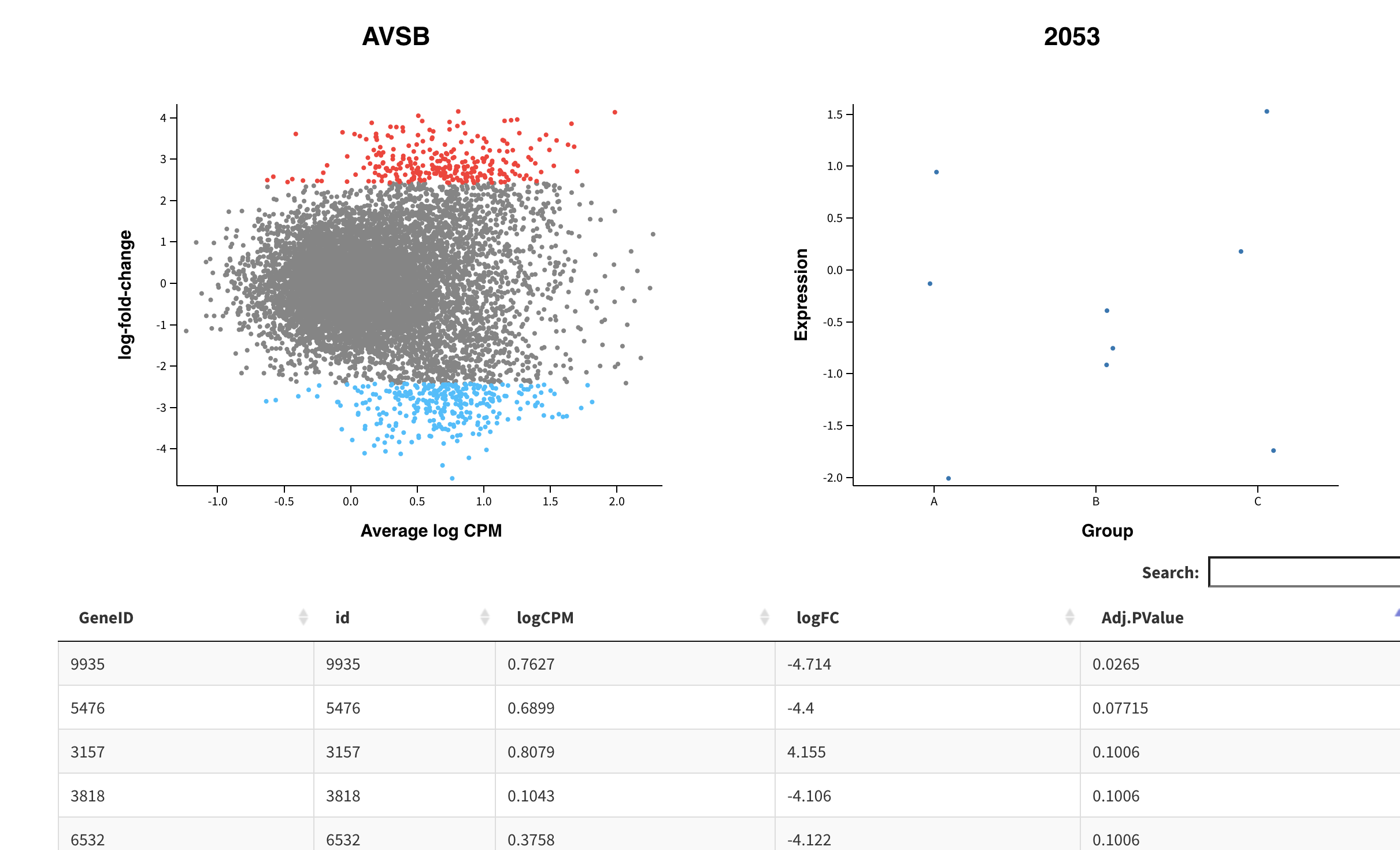
赶快试试吧,把你以前的转录组数据处理项目都可以使用这个Glimma 做出来一个交互式可视化RNA-seq数据网页报告哦!
如果是真实转录组数据直接使用示例代码即可
我上面演示的是自己创造的一个表达量矩阵,具体见生信技能树公众号教程:3个分组的表达量矩阵的两两之间差异分析,但是如果你有真实的数据:
- https://bioconductor.org/packages/release/bioc/vignettes/Glimma/inst/doc/DESeq2.html
- https://bioconductor.org/packages/release/bioc/vignettes/Glimma/inst/doc/limma_edger.html
library(Glimma)
library(edgeR)
library(DESeq2)
dge <- readRDS(system.file("RNAseq123/dge.rds", package = "Glimma"))
dds <- DESeqDataSetFromMatrix(
countData = dge$counts,
colData = dge$samples,
rowData = dge$genes,
design = ~group
)
glimmaMDS(dds)
dds <- DESeq(dds, quiet=TRUE)
glimmaMA(dds)
另外:上面的代码大量涉及到R基础知识:
需要把R的知识点路线图搞定,如下:
- 了解常量和变量概念
- 加减乘除等运算(计算器)
- 多种数据类型(数值,字符,逻辑,因子)
- 多种数据结构(向量,矩阵,数组,数据框,列表)
- 文件读取和写出
- 简单统计可视化
- 无限量函数学习