传统的甲基化信号值通常是芯片,比如illumina公司的27K,450K,850K的甲基化芯片,它们检查的通常是细胞混合物,比如绝大部分肿瘤组织样品其实是混合了上皮细胞,基质细胞和免疫细胞。如果我们感兴趣肿瘤组织的纯度,或者说肿瘤的免疫浸润情况,就可以使用同样的反卷积方法来。这个时候EpiDISH包就可以派上用场:
集成3大算法
EpiDISH 包的例子倾向于使用 RPC 方法,全部的3个算法是:
- (Robust Partial Correlations-RPC(Teschendorff et al. 2017),
- Cibersort-CBS(Newman et al. 2015),
- Constrained Projection-CP(Houseman et al. 2012)),
自带4个参考数据集
看 BMC Bioinformatics 发表于2017的文章:《A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies 》,里面有详细介绍这4个参考数据集如何制作的
- two whole blood subtypes reference,
- one generic epithelial reference with epithelial cells, fibroblasts, and total immune cells,
- one reference for breast tissue
可以很容易看到这些参考数据集都被打包好了,data(package = ‘EpiDISH’)
Data sets in package ‘EpiDISH’:
DummyBeta.m Dummy beta value matrix
LiuDataSub.m Whole blood example beta value matrix
centBloodSub.m Whole blood reference of 188 tsDHS-DMCs and 7 blood
cell subtypes
centDHSbloodDMC.m Whole blood reference of 333 tsDHS-DMCs and 7 blood
cell subtypes
centEpiFibFatIC.m Reference for breast tissue
centEpiFibIC.m Reference for genenric epithelial tissue
我们首先看看上皮细胞的参考数据集,在EpiDISH 包给命名为 centEpiFibIC.m
library(EpiDISH)
data(centEpiFibIC.m)
# 可以看到是3个细胞类型的716个甲基化探针
dim(centEpiFibIC.m)
head(centEpiFibIC.m)
pheatmap::pheatmap(centEpiFibIC.m,
show_rownames = F)
可以看到是每个细胞亚群都有自己的特异性甲基化芯片探针:
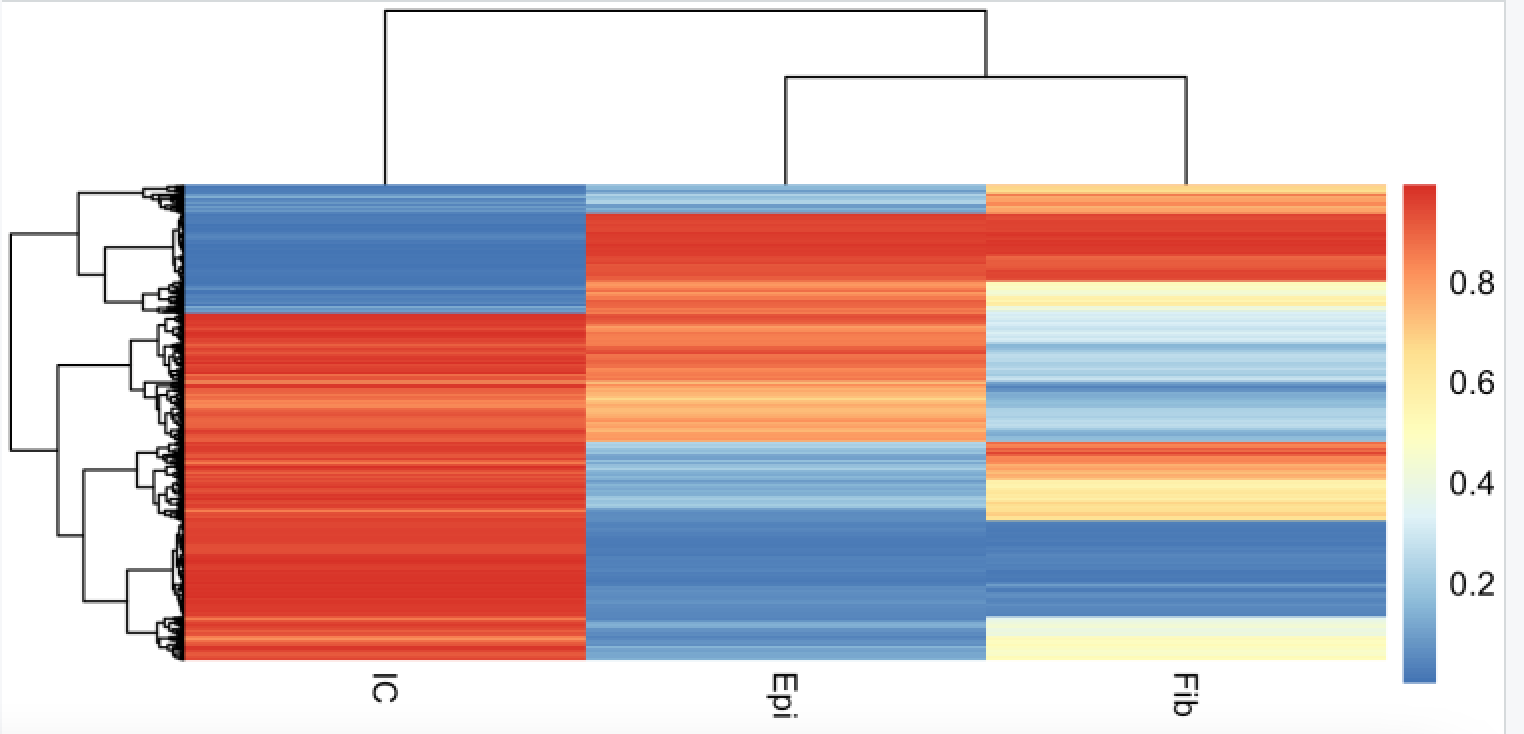
感兴趣的可以去看文献,这些探针是如何得到的。
> dim(centEpiFibIC.m)
[1] 716 3
> head(centEpiFibIC.m)
Epi Fib IC
cg16509569 0.97905062 0.97827048 0.006030438
cg05596756 0.97643990 0.98184238 0.006086343
cg11802899 0.97407930 0.97413265 0.006219681
cg02026204 0.96812178 0.97424952 0.005418776
cg16622899 0.01658659 0.01585706 0.984393223
cg24002183 0.02297836 0.02369267 0.975103840
再看看 其中一个血液组成评价数据集,centDHSbloodDMC.m
> data(centDHSbloodDMC.m)
> dim(centDHSbloodDMC.m)
[1] 333 7
> head(centDHSbloodDMC.m)
B NK CD4T CD8T Mono Neutro Eosino
cg01024458 0.034 0.962 0.969 0.958 0.987 0.992 0.991
cg11661493 0.037 0.954 0.971 0.964 0.958 0.962 0.962
cg21596498 0.060 0.980 0.989 0.989 0.980 0.976 0.984
cg05205074 0.049 0.954 0.967 0.961 0.967 0.979 0.986
cg17232476 0.045 0.962 0.969 0.958 0.932 0.980 0.976
cg14936008 0.065 0.976 0.980 0.983 0.977 0.983 0.986
> pheatmap::pheatmap(centDHSbloodDMC.m,show_rownames = F)
可以看到也是基本上符合认知,NK,CD4,CD8会比较接近,都是T细胞。
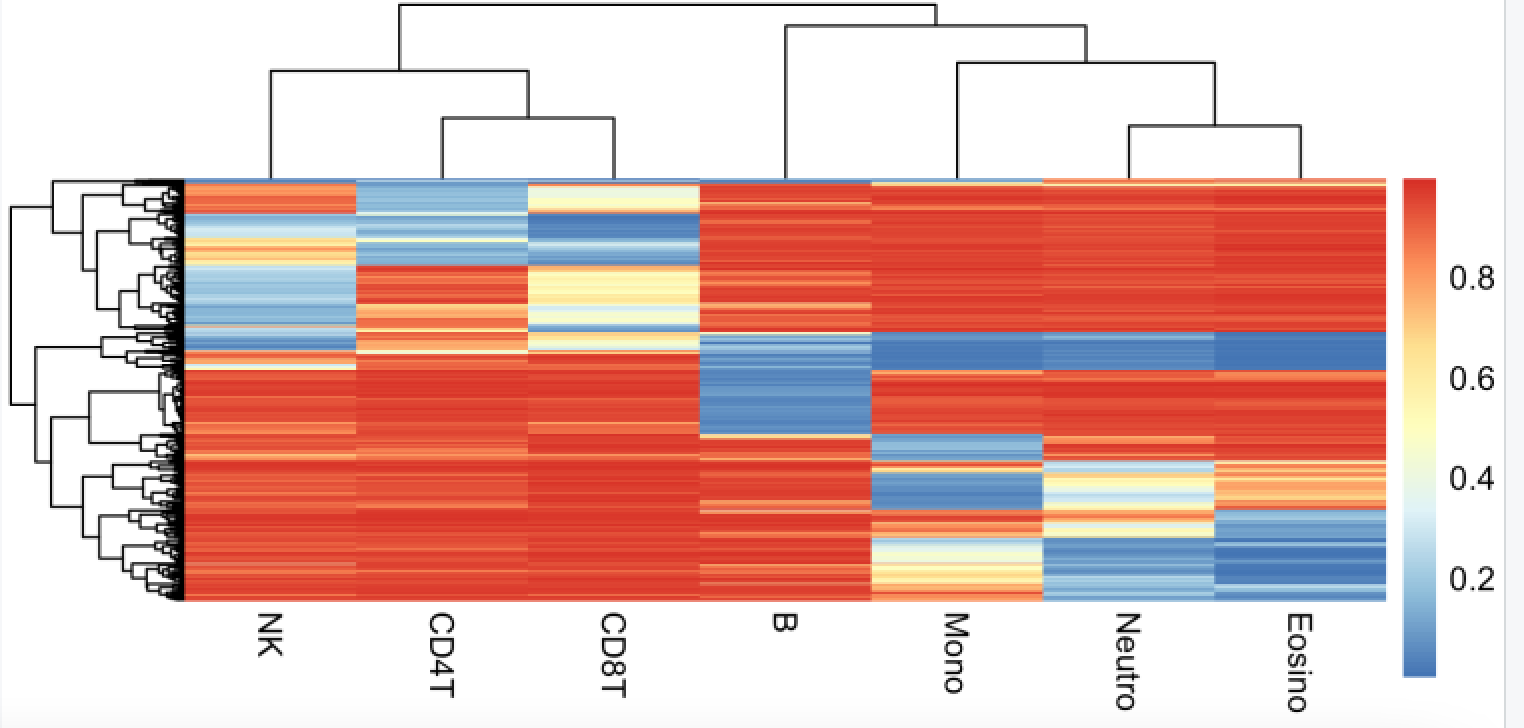
上皮细胞混合物的分解
使用起来非常简单,就是 epidish 函数调用即可,它需要一个甲基化信号值矩阵,如下所示:
data(DummyBeta.m)
# 可以看到是10个甲基化样品,每个都是节选了 2000个甲基化探针
dim(DummyBeta.m)
DummyBeta.m[1:4,1:4]
out.l <- epidish(beta.m = DummyBeta.m,
ref.m = centEpiFibIC.m,
method = "RPC"
)
out.l$estF
# 各个样品的各个细胞比例
pheatmap::pheatmap(out.l$estF)
rowSums(out.l$estF)
3种细胞比例之和在每个样品都是1,可以看到10个样品各自的细胞组成差异很大:
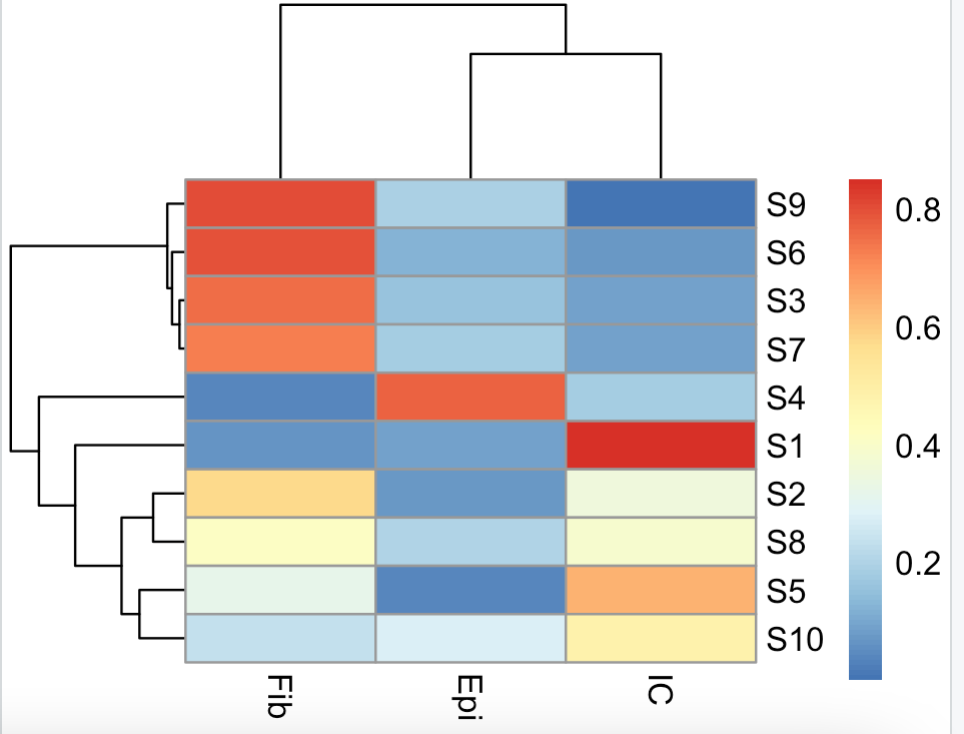
免疫细胞混合物的分解
使用起来非常简单,就是 epidish 函数调用即可,它需要一个甲基化信号值矩阵,如下所示:
data(LiuDataSub.m)
BloodFrac.m <- epidish(beta.m = LiuDataSub.m,
ref.m = centDHSbloodDMC.m,
method = "RPC")$estF
head(BloodFrac.m)
pheatmap::pheatmap(BloodFrac.m)
boxplot(BloodFrac.m)
可以看到绝大部分样品都是以 Neutro 为主,占比超80% ,剩余的6种免疫细胞比例含量都很低:
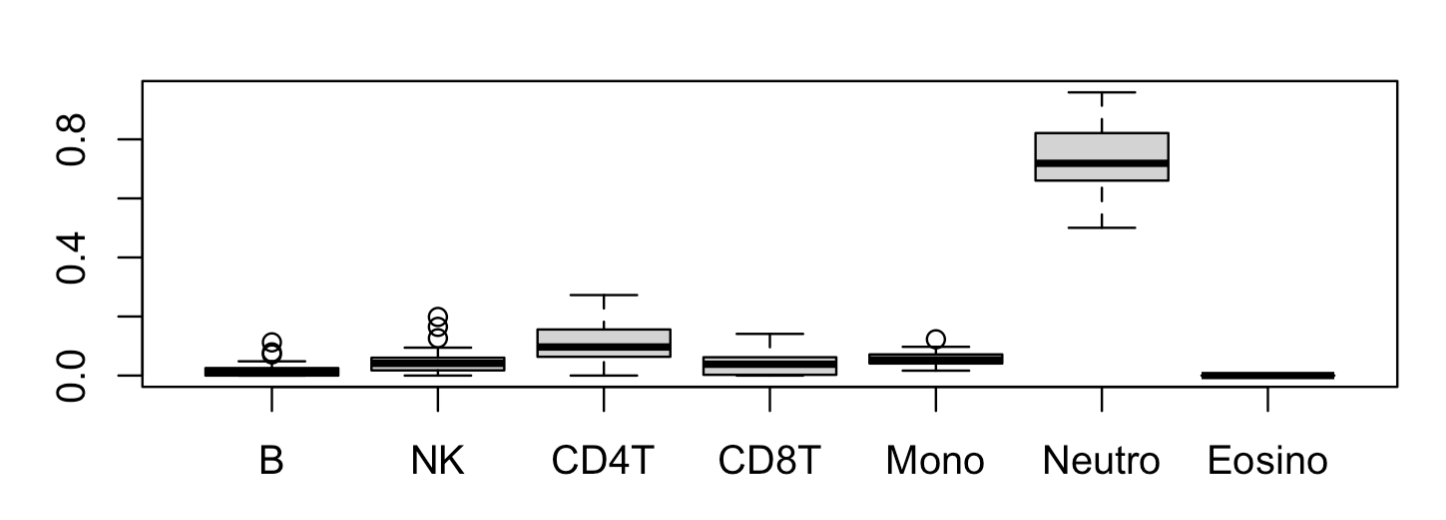
高级函数hepidish做两次分解
第一次分解可以把肿瘤样品区分成为上皮细胞,基质细胞和免疫细胞,第二次分解就针对免疫细胞进行细分亚群,使用方法如下:
data(centBloodSub.m)
frac.m <- hepidish(beta.m = DummyBeta.m,
ref1.m = centEpiFibIC.m,
ref2.m = centBloodSub.m[,c(1, 2, 5)],
h.CT.idx = 3, method = 'RPC')
frac.m
boxplot(frac.m)
这个其实挺适合肿瘤免疫微环境的分解,值得试试看!
最高级用法 CellDMC
这个看起来是做Epigenome-Wide Association Studies(EWAS),有点超纲了,下次再讲解!
甲基化背景知识
甲基化测序的 WGBS和RRBS,还有 芯片是最高频的甲基化技术,其中甲基化芯片数据处理我是有视频课程的,首先需要阅读我在生信技能树的甲基化系列教程,目录如下:
- 01-甲基化的一些基础知识.pdf
- 02-甲基化芯片的一般分析流程.pdf
- 03-甲基化芯片数据下载的多种技巧.pdf
- 04-甲基化芯片数据下载如何读入到R里面.pdf
- 05-甲基化芯片数据的一些质控指标.pdf
- 06-甲基化信号值矩阵差异分析哪家强.pdf
- 07-甲基化芯片信号值矩阵差异分析的标准代码.pdf
- 08-TCGA数据库的各个癌症甲基化芯片数据重新分析.pdf
- 09-TCGA数据库的癌症甲基化芯片数据重分析.pdf
- 10-TCGA数据辅助甲基化区域的功能研究.pdf
- 11-按基因在染色体上的顺序画差异甲基化热图.pdf
- 850K甲基化芯片数据的分析.pdf
- 使用DSS包多种方式检验差异甲基化信号区域.pdf
然后就可以看我在B站免费分享的视频课程《甲基化芯片(450K或者850K)数据处理 》
肿瘤免疫细胞比例推断背景知识
肿瘤微环境是肿瘤细胞生存和发展的土壤,其中浸润到肿瘤局部的免疫细胞介导了肿瘤免疫微环境,tumor immune microenvironment, 简称TIME。肿瘤的免疫疗法作用于免疫微环境, 肿瘤免疫微环境的异质性与免疫疗法的不同响应率必然存在这密切的关系,免疫微环境有望作为一种biomaker来指导临床治疗,筛选能够从免疫疗法中获益的肿瘤患者群体。
研究肿瘤免疫微环境组成的技术手段有很多,基于测序技术和生物信息学分析的方法是目前较为流程的一种策略。通过转录组测序,可以获得肿瘤样本中不同基因的表达量数据,通过对应的生物信息学软件,可以得到肿瘤免疫微环境中各种细胞的表达量,从而对肿瘤微环境进行分型,识别浸润的免疫细胞亚群,比较不同亚群的表达情况,结合生存分析,进一步筛选某种微环境亚型或者免疫细胞亚群作为biomarker。
基于转录组的算法策略
基于基因表达谱数据,分析肿瘤免疫微环境组分的软件有很多,大致分成了以下3种策略
1. marker gene
将不同免疫细胞对应的marker genes作为基因集合, 采用类似GSEA的算法来评估样本中高表达的基因在不同免疫细胞的基因集合中是否富集,该策略对应的软件如下
- TIminer
- xCell
- MCP-counter
2. 反卷积
卷积和反卷积是深度学习中常见的算法,将每个样本看做是多种免疫细胞的混合,采用线性回归拟合出每种免疫细胞的组分和表达量与最终混合后的关系,通过反卷积算法,提取每种免疫细胞的表达特征,该策略对应的软件如下
- CIBERSORT
- TIMER
- EPIC
- quanTIseq
3. NMF非负矩阵分解
类似突变特征分析,NMF算法也可以用于从基因表达谱中提取免疫细胞表达谱特征,该策略对应的软件如下
- deconf
- ssKL
- ssFrobenius
参考文献:
\1. Hackl H, Charoentong P, Finotello F, et al. Computational genomics tools for dissecting tumour-immune cell interactions. Nat Rev Genet 2016;17:441-58.
\2. Finotello, Francesca, and Zlatko Trajanoski. Quantifying Tumor-Infiltrating Immune Cells from Transcriptomics Data. Cancer Immunology, Immunotherapy: CII 67, no. 7 (2018): 1031–40.