ATAC-seq的数据分析主要是检测信号峰值,就是peaks,不同样品的peaks的差异主要是两个思路,使用韦恩图展现有无peaks的差异,另外就是使用散点图展现高低强弱的peaks差异。
现在是2021了,有了很多成熟的软件算法可以做peaks的差异分析,不过偶尔忆苦思甜也是有必要的ATAC-seq经典差异分析,让我们一起看看距离2013年的ATAC-seq技术开发出来不到两年的 2015的一个文章是如何做。文章是:A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol 2015 Dec 18;16:284. PMID: 26683334
数据集在:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67382
可以看到里面的ATAC-seq的数据是4个:
GSM1645706 BAFi_ATAC_rep1
GSM1645707 BAFi_ATAC_rep2
GSM1645708 CTRL_ATAC_rep1
GSM1645709 CTRL_ATAC_rep2
可以看到很明显的2个组,每个组都是2个重复的样品,而且根据peaks的信号值强度相关性散点图可以看出来组内一致性比较好:
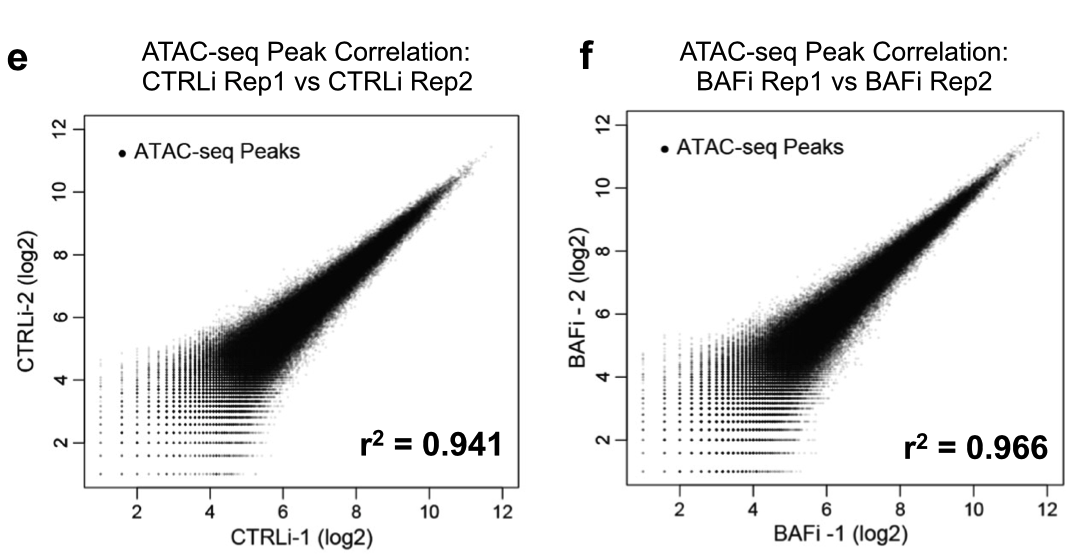
其实现在有Irreproducibility Discovery Rate (IDR)指标,用于评估重复样本间peaks一致性。IDR评估会同时考虑peaks间的overlap和富集倍数的一致性。通过IDR阈值(0.05)的占比越大,说明重复样本间peaks一致性越好。
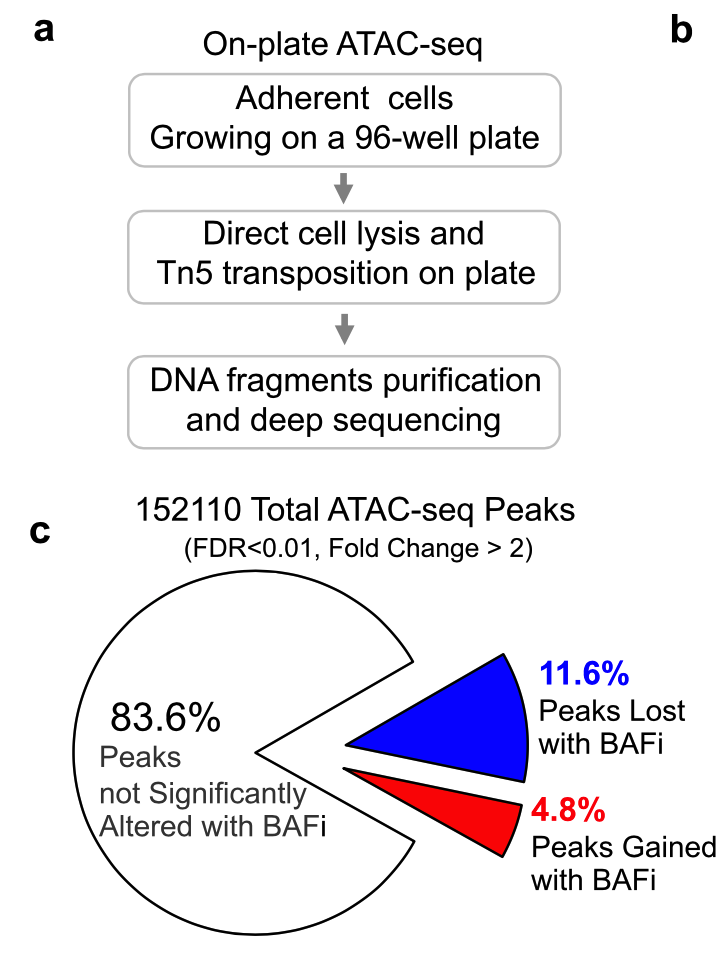
差异分析主居然还像模像样的给出来了一个流程图,简单的上下调的peaks数量,还可以画一个饼图:
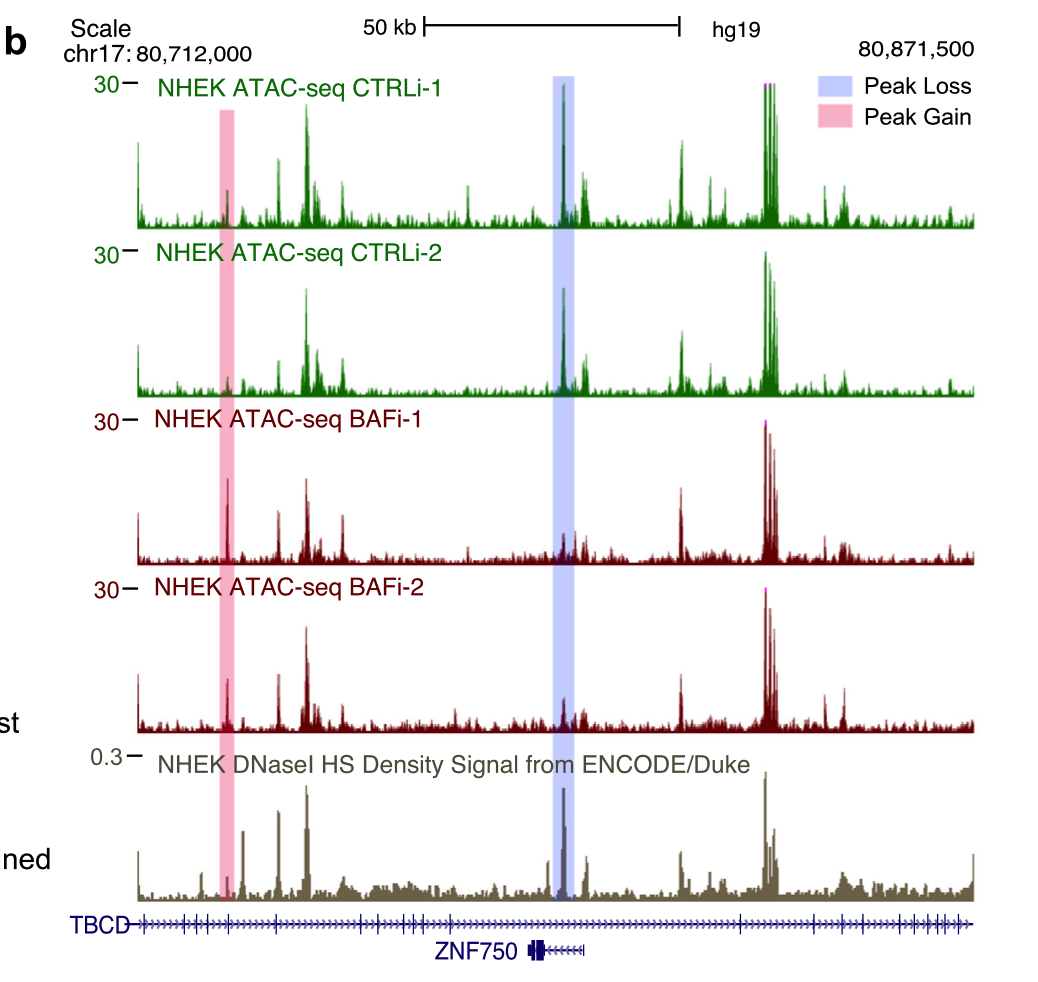
挑选具体的基因,进行IGV软件载入bw文件的可视化,看ATAC-seq的信号差异:
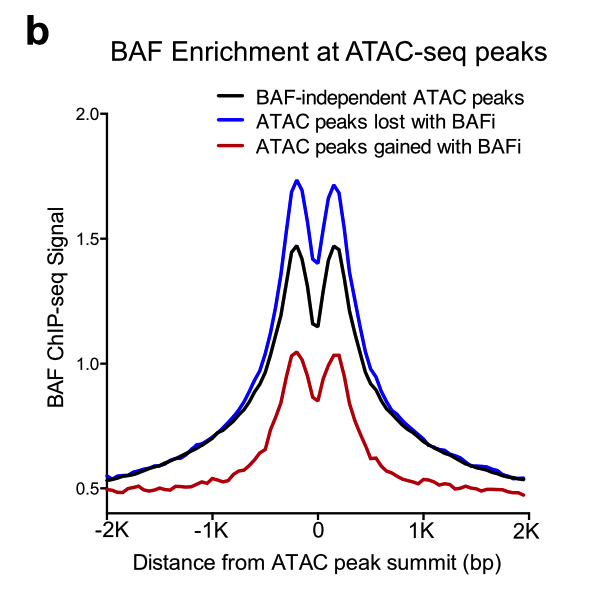
另外一个不得不提的经典图表,就是看信号强度的:
处理流程
首先看上游数据处理流程:
- ATAC-seq paired-end reads were trimmed for Illumina adapter sequences using a custom script
- mapped to hg19 using bowtie with parameters –S –X2000 –m1.
- Duplicate reads were discarded with samtools.
- Peaks were identified using macs2 with parameters callpeak –nomodel –shift -100 –extsize 200.
- Peak sets called in individual replicates were combined and individual peaks merged if overlapping within 300 bp to form a union peak set.
下游就是差异分析; - Differentially accessible peaks from this union set were identified with edgeR by counting all read ends overlapping peaks in each condition.
- edgeR was run with default settings, a fold-change threshold of 2, and FDR < 0.01.
其实呢,现在的ATAC-seq 数据处理的更完善了,见ATAC-seq项目的标准分析仅收费1600,差异分析也有专门的R包,比如 Diffbind,有一个2020综述《From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis》值得看: - MACS2 进行 Peak calleing
- csaw 进行差异 Peak 分析
- MEME suite 进行 motif 检测和富集
- ChIPseeker 进行注释和可视化
- HMMRATAC 进行核小体检测
- HINT-ATAC 进行足迹分析
结合转录组测序
可以看到是3个分组,共6个样品:
GSM1921004 Ctrli_rep1_RNAseq GSM1921005 Ctrli_rep2_RNAseq GSM1921006 BAFi_rep1_RNAseq GSM1921007 BAFi_rep2_RNAseq GSM1921008 p63i_rep1_RNAseq GSM1921009 p63i_rep2_RNAseq两次差异分析,各自挑选 (fold change > 3 with depletion, FDR < 0.01)的基因,然后看交集:
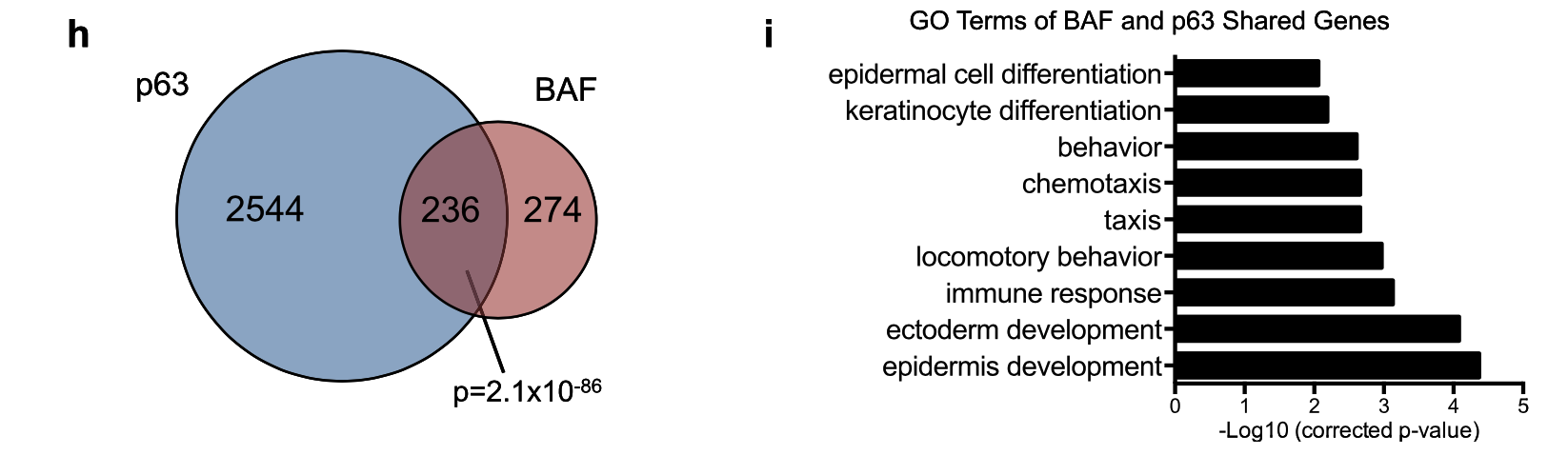
对 236 genes controlled jointly by both BAF and p63 进行GO数据库的功能注释。
这个转录组数据分析比较容易复现,基本上看我六年前的表达芯片的公共数据库挖掘系列推文即可; - 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够