发表这个算法的文章是2012年:《Camera: a competitive gene set test accounting for inter-gene correlation》,链接是:https://academic.oup.com/nar/article/40/17/e133/2411151 如果是第一次接触生物信息学软件的小伙伴可能是会觉得有点老。

而且呢,里面的公式一大把,看起来会比较头疼,其实可以把它理解为 gsea的类似方法即可,gsea分析这方面教程我在《生信技能树》公众号写了不少了,不管是芯片还是测序的表达矩阵,都是一样的,把全部基因排序即可:
- 比如你有2万个基因,你根据自己的条件分组后算差异情况,根据差异把基因排序,然后看缺氧相关200个基因组成的集合在全部的排好序的2万个基因是散乱分布,还是集中于头部和尾部。
- 当然了,基因集肯定不仅仅是缺氧这个生物学功能啦,在msigdb数据库有几万基因集合,其实生物学背景更重要。
- 另外,基因的排序也不仅仅是条件分组后算差异来排序,也可以仅仅是表达量高低排序。
目前这个 camera 功能实现起来非常的方便,因为它仅仅是limma包的一个函数:camera: Competitive Gene Set Test Accounting for Inter-gene Correlation
首先呢,我们认为的创造一个表达量矩阵,1000行代表1000个基因,然后6个样品区分成为2个组,这样的话可以很方便做差异分析:
library(limma)
y <- matrix(rnorm(1000*6),1000,6)
design <- cbind(Intercept=1,Group=c(0,0,0,1,1,1))
design
# First set of 20 genes are genuinely differentially expressed
index1 <- 1:20
y[index1,4:6] <- y[index1,4:6]+1
# Second set of 20 genes are not DE
index2 <- 21:40
可以看到,我们把前面的20个基因整体呢在右边的3个样品表达量增加了1,所以前面的20个基因组成的集合理论上应该是功能富集啦。但是index2 <- 21:40代表的基因集我们并没有处理它,它就是一个随机数,所以理论上不应该是被富集。
接下来就使用limma包的一个函数:camera: Competitive Gene Set Test Accounting for Inter-gene Correlation 对这两个基因集进行统计学检验吧:
> camera(y, index1, design)
NGenes Direction PValue
set1 20 Up 3.110234e-10
> camera(y, index2, design)
NGenes Direction PValue
set1 20 Up 0.9888623
>
>
> camera(y, list(set1=index1,set2=index2),
+ design, inter.gene.cor=NA)
NGenes Correlation Direction PValue FDR
set1 20 -0.01269341 Up 0.002283103 0.004566206
set2 20 -0.02253970 Up 0.988594938 0.988594938
> camera(y, list(set1=index1,set2=index2),
+ design, inter.gene.cor=0.01)
NGenes Direction PValue FDR
set1 20 Up 3.110234e-10 6.220468e-10
set2 20 Up 9.888623e-01 9.888623e-01
可以看到,只有第一个被我们认为干扰的基因集,被认为是统计学显著的上升。
同样的,我们也可以很方便的可视化一下:
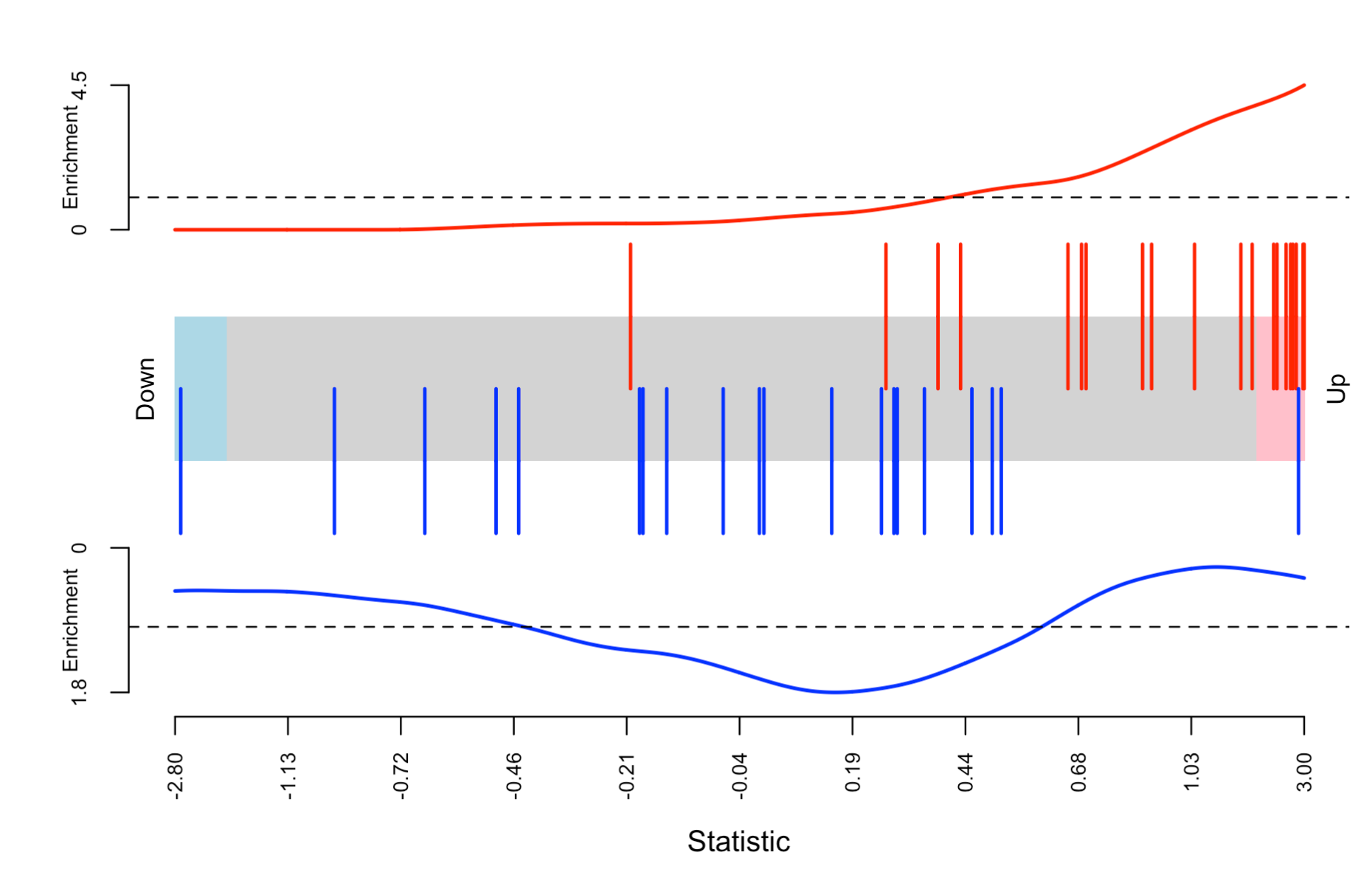
stat=rowMeans(y[,4:6]) - rowMeans(y[,1:3])
# One directional
barcodeplot(stat, index = index1)
# Two directional
barcodeplot(stat, index = index1,
index2 = index2)
如下所示:
上面的代码大量涉及到R基础知识:
需要把R的知识点路线图搞定,如下:
- 了解常量和变量概念
- 加减乘除等运算(计算器)
- 多种数据类型(数值,字符,逻辑,因子)
- 多种数据结构(向量,矩阵,数组,数据框,列表)
- 文件读取和写出
- 简单统计可视化
- 无限量函数学习