gsea分析这方面教程我在《生信技能树》公众号写了不少了,不管是芯片还是测序的表达矩阵,都是一样的,把全部基因排序即可:
- 比如你有2万个基因,你根据自己的条件分组后算差异情况,根据差异把基因排序,然后看缺氧相关200个基因组成的集合在全部的排好序的2万个基因是散乱分布,还是集中于头部和尾部。
- 当然了,基因集肯定不仅仅是缺氧这个生物学功能啦,在msigdb数据库有几万基因集合,其实生物学背景更重要。
- 另外,基因的排序也不仅仅是条件分组后算差异来排序,也可以仅仅是表达量高低排序。
msigdb数据库网页里面有着丰富的基因集,MSigDB(Molecular Signatures Database)数据库中定义了已知的基因集合:http://software.broadinstitute.org/gsea/msigdb 包括H和C1-C7八个系列(Collection),每个系列分别是:
- H: hallmark gene sets (癌症)特征基因集合,共50组,最常用;
- C1: positional gene sets 位置基因集合,根据染色体位置,共326个,用的很少;
- C2: curated gene sets:(专家)校验基因集合,基于通路、文献等:
- C3: motif gene sets:模式基因集合,主要包括microRNA和转录因子靶基因两部分
- C4: computational gene sets:计算基因集合,通过挖掘癌症相关芯片数据定义的基因集合;
- C5: GO gene sets:Gene Ontology 基因本体论,包括BP(生物学过程biological process,细胞原件cellular component和分子功能molecular function三部分)
- C6: oncogenic signatures:癌症特征基因集合,大部分来源于NCBI GEO 发表芯片数据
- C7: immunologic signatures: 免疫相关基因集合。
如下所示的一个经典的gsea图:
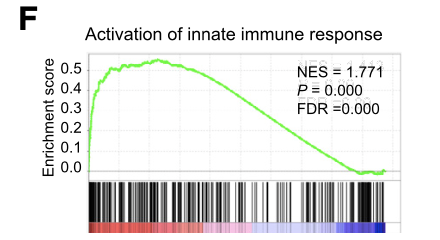
它就很好的说明了这个 innate immune response基因集,是在排序好的2万多个基因里面是左偏的,也就是说这个通路是激活的。
但是这个gsea分析对绝大部分初学者来说理解起来很困难,而且代码实现难度也不小。最近看教程,发现其实limma包就有一个barcodeplot函数,其示例代码很容易让人理解:
library(limma)
stat <- rnorm(100)
fivenum(stat)
sel <- 1:10
sel2 <- 11:20
stat[sel] <- stat[sel]+1
stat[sel2] <- stat[sel2]-1
stat
plot(stat)
首先呢,上面的代码创造了一个向量,是100个数值,它们最开始时是乱序,但是我们人为的把前面的10个数值加上了1,这样前面的10个数值就会名列前茅。然后呢,我们人为的把第11到20个数值减去1,这样的它们数值会偏小,但是并不会垫底。
下面的barcodeplot可视化你的基因排序 ,很容易看出来:
# One directional
barcodeplot(stat, index = sel)
# Two directional
barcodeplot(stat, index = sel, index2 = sel2)
可以看到,第一个基因集合, 就是前面的10个数值其实是遥遥领先,而第二个基因集合,就是第11到20个数值会比较小,但不会是绝对的垫底。
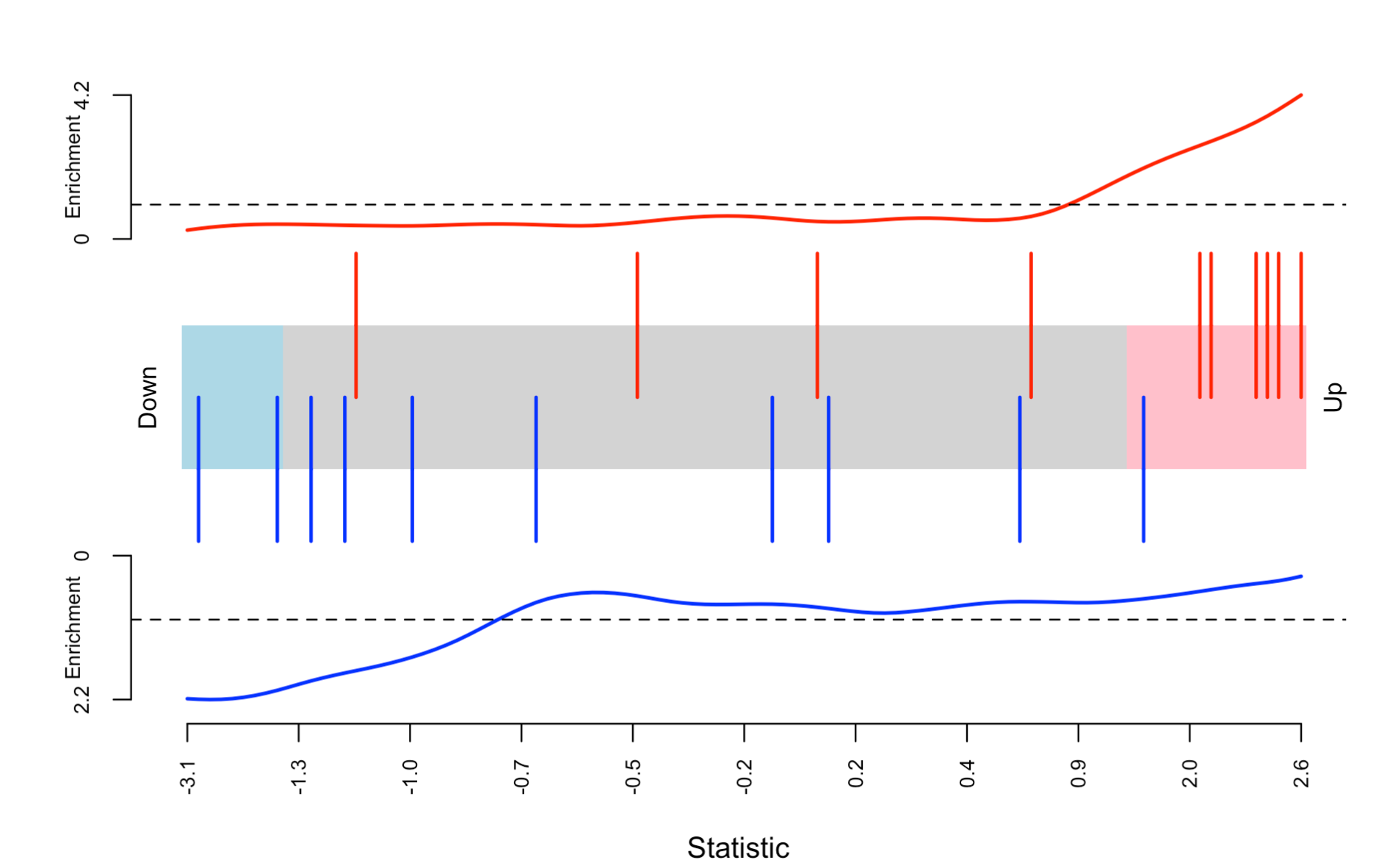
亲爱的读者,发挥你聪明的小脑瓜,思考一下,假如你在前面把 人为的把第11到20个数值减去10,这样的话,第二个基因集合,是不是就可以看到很明显的垫底情况了?
上面的代码大量涉及到R基础知识:
需要把R的知识点路线图搞定,如下:
- 了解常量和变量概念
- 加减乘除等运算(计算器)
- 多种数据类型(数值,字符,逻辑,因子)
- 多种数据结构(向量,矩阵,数组,数据框,列表)
- 文件读取和写出
- 简单统计可视化
- 无限量函数学习