很多时候,我们都没办法很快判断seurat默认聚类分群后的每个亚群的生物学命名,会短暂的把大家先归纳为一个大类,比如肿瘤单细胞数据第一次分群通用规则,按照 :
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibo or CD31+,PECAM1,endo)
这3大亚群都有自己的标记基因,它们其实都是涵盖了非常多的亚群,这个时候就需要一定程度的代码进行合并它们多个单细胞亚群。
下面我们以 seurat 官方教程为例:
rm(list = ls())
library(Seurat)
# devtools::install_github('satijalab/seurat-data')
library(SeuratData)
library(ggplot2)
library(patchwork)
library(dplyr)
load(file = 'basic.sce.pbmc.Rdata')
DimPlot(pbmc, reduction = 'umap',
label = TRUE, pt.size = 0.5) + NoLegend()
sce=pbmc
如果你不知道 basic.sce.pbmc.Rdata 这个文件如何得到的,麻烦自己去跑一下 可视化单细胞亚群的标记基因的5个方法,自己 save(pbmc,file = ‘basic.sce.pbmc.Rdata’) ,我们后面的教程都是依赖于这个 文件哦!
首先查看标记基因
其实缺一个高质量非冗余单细胞亚群标记基因数据库,假如我们的生物学认知不够,就不需要把T细胞分成 “Naive CD4 T” , “Memory CD4 T” , “CD8 T”, “NK” 这些亚群,可以合并为T细胞这个大的亚群:
# 参考;https://mp.weixin.qq.com/s/9d4X3U38VuDvKmshF2OjHA
genes_to_check = c('PTPRC', 'CD3D', 'CD3E', 'PRF1' , 'NKG7',
'CD19', 'CD79A', 'MS4A1' ,
'CD68', 'CD163', 'CD14',
"FCER1A", "PPBP")
DotPlot(sce, group.by = 'seurat_clusters',
features = unique(genes_to_check)) + RotatedAxis()
如果生物学背景知识不够,也可以勉强把细胞亚群进行生物学命名:
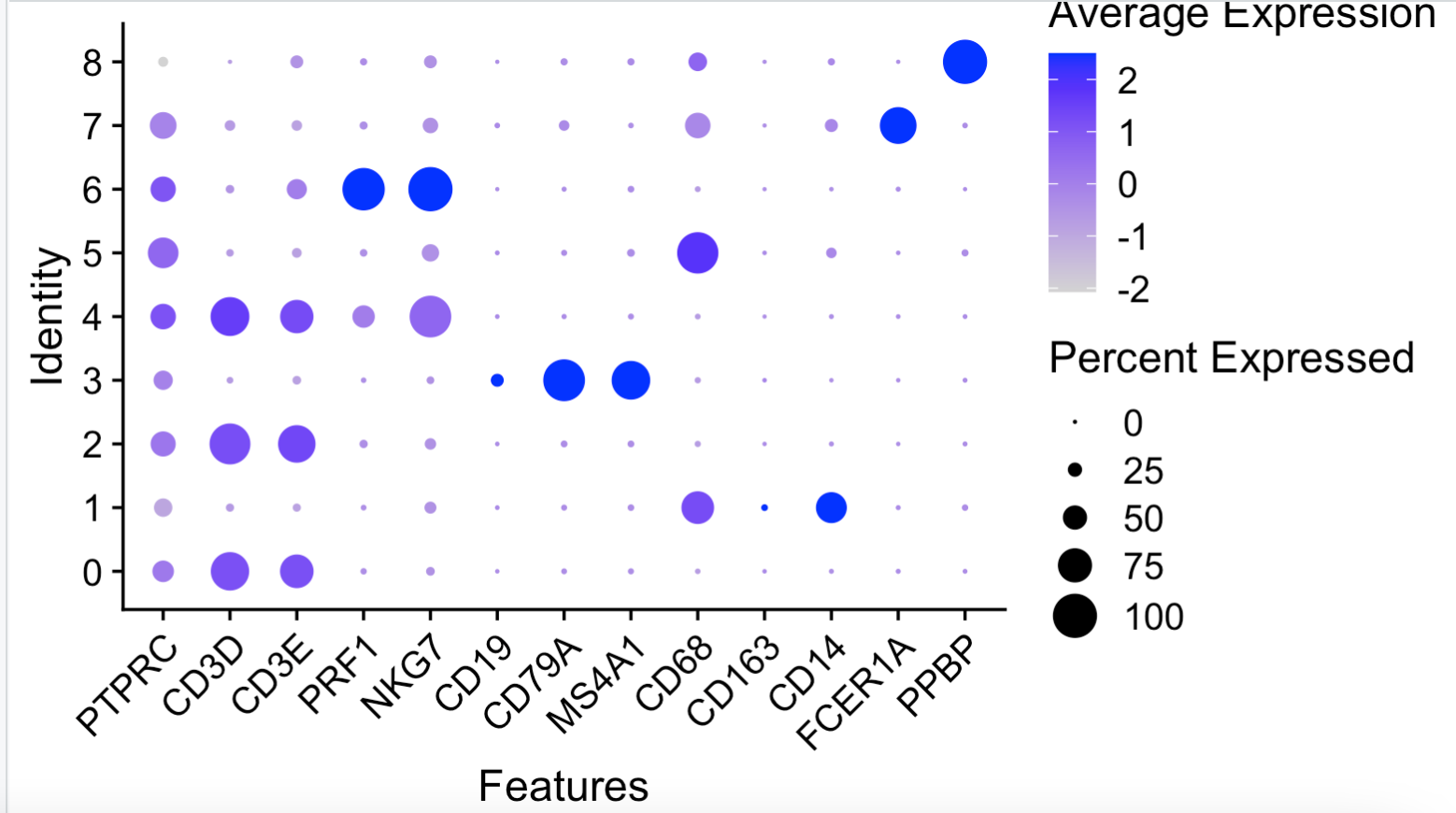
我们这里假装自己的生物学背景很弱,所以只能是区分 B DC Mono Platelet T 这5个细胞亚群。
方法一:使用 RenameIdents 函数
Idents(sce)
levels(sce)
head(sce@meta.data)
# method : 1
new.cluster.ids <- c("T", "Mono", "T",
"B", "T", "Mono",
"T", "DC", "Platelet")
names(new.cluster.ids) <- levels(sce)
sce <- RenameIdents(sce, new.cluster.ids)
DimPlot(sce, reduction = 'umap',
label = TRUE, pt.size = 0.5) + NoLegend()
方法二:使用unname函数配合向量:
cluster2celltype <- c("0"="T",
"1"="Mono",
"2"="T",
"3"= "B",
"4"= "T",
"5"= "Mono",
"6"= "T",
"7"= "DC",
"8"= "Platelet")
sce[['cell_type']] = unname(cluster2celltype[sce@meta.data$seurat_clusters])
DimPlot(sce, reduction = 'umap', group.by = 'cell_type',
label = TRUE, pt.size = 0.5) + NoLegend()
方法三:使用数据框
# 一个数据框
(n=length(unique(sce@meta.data$seurat_clusters)))
celltype=data.frame(ClusterID=0:(n-1),
celltype='unkown')
celltype[celltype$ClusterID %in% c(0,2,4,6),2]='T'
celltype[celltype$ClusterID %in% c(3),2]='B'
celltype[celltype$ClusterID %in% c(1,5),2]='Mono'
celltype[celltype$ClusterID %in% c(7),2]='DC'
celltype[celltype$ClusterID %in% c(8),2]='Platelet'
sce@meta.data$celltype = "NA"
for(i in 1:nrow(celltype)){
sce@meta.data[which(sce@meta.data$seurat_clusters == celltype$ClusterID[i]),'celltype'] <- celltype$celltype[i]}
table(sce@meta.data$celltype)
DimPlot(sce, reduction = 'umap', group.by = 'celltype',
label = TRUE, pt.size = 0.5) + NoLegend()
head(sce@meta.data)
table(sce@meta.data$cell_type,
sce@meta.data$celltype)
可以看到多种方法都是一样的效果:
B DC Mono Platelet T
B 342 0 0 0 0
DC 0 32 0 0 0
Mono 0 0 642 0 0
Platelet 0 0 0 14 0
T 0 0 0 0 1608
得到了如下所示的粗糙分群:
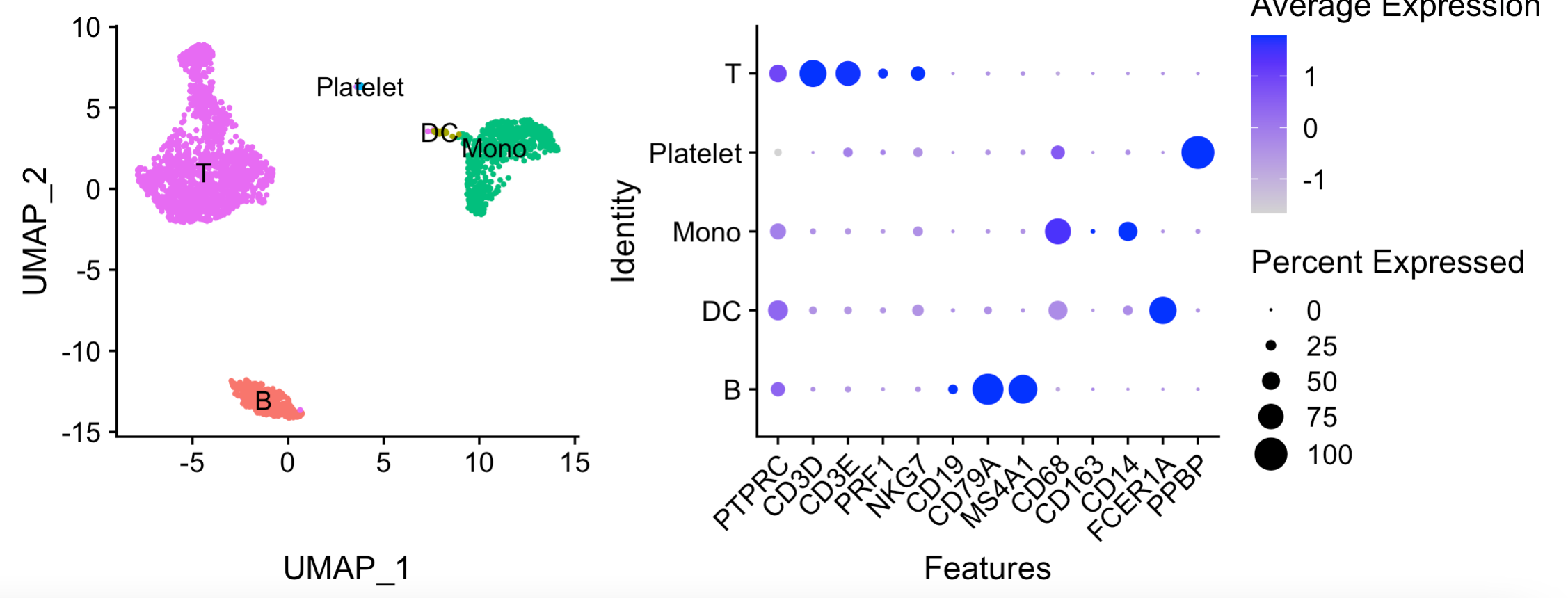
绘图代码如下:
p1=DimPlot(sce, reduction = 'umap', group.by = 'celltype',
label = TRUE, pt.size = 0.5) + NoLegend()
p2=DotPlot(sce, group.by = 'celltype',
features = unique(genes_to_check)) + RotatedAxis()
library(patchwork)
p1+p2