疫情期间发布了《肿瘤基因测序数据分析》课程,:
目录如下:
基于Linux的上游数据分析流程
质量控制
比对
GATK流程
mutect流程
CNV流程
基于R语言的下游统计可视化
突变全景图
SNV
CNV(GISTIC)
临床
pathway等等
基因排序,样本排序,突变分类,突变总结
多个队列(癌症,亚型,复发与否,转移与否)
特定基因集突变图
生存
棒棒糖图
3种pathway展示图
突变频谱
6碱基
96碱基
signature(COSMIC reference)
NMF(自己构建)
视频免费发放在b站,学习方式如下所示:
因为使用的是百度李彦宏的文章数据,大家会比较倾向于处理tcga的肿瘤突变数据,虽然仅仅是输入数据的不一样,后续分析都是靠 maftools 这个包,maftools 全能无需我再吹嘘,必须花十几个小时认真掌握它!
假如大家是在 https://xenabrowser.net/datapages/ ,找到 GDC TCGA Breast Cancer (BRCA) (20 datasets) ,数据库提供了4种somatic突变的maf文件供下载,somatic mutation (SNPs and small INDELs) ,一般来说我们选择GATK团队出品的MuTect2 软件拿到的somatic突变数据文件;
- MuTect2 Variant Aggregation and Masking (n=986) GDC Hub
这个时候呢,你会发现下载的突变数据是tsv格式,并不是maf格式,读入这样的tsv格式的肿瘤突变是信息需要一定技巧哦!
Data from different samples is combined into mutationVector; "Hugo_Symbol", "Chromosome", "Start_Position", "End_Position", "Reference_Allele", "Tumor_Seq_Allele2", "Tumor_Sample_Barcode", "HGVSp_Short" and "Consequence" data are renamed accordingly and presented; "dna_vaf" data is added and is calculated by "t_alt_count"/"t_depth".
如下所示的文件;
https://gdc-hub.s3.us-east-1.amazonaws.com/download/TCGA-BRCA.mutect2_snv.tsv.gz
如果我们仅仅是读入 TCGA-BRCA.mutect2_snv.tsv.gz,会发现它没办法导入到maftools包。
我简单的修改了一下读入方式,代码如下:
rm(list = ls())
require(maftools)
options(stringsAsFactors = F)
library(data.table)
tmp=fread('TCGA-BRCA.mutect2_snv.tsv.gz')
head(tmp)
colnames(tmp) =c( "Tumor_Sample_Barcode", "Hugo_Symbol",
"Chromosome", "Start_Position",
"End_Position", "Reference_Allele", "Tumor_Seq_Allele2",
"HGVSp_Short" , 'effect' ,"Consequence",
"vaf" )
tmp$Entrez_Gene_Id =1
tmp$Center ='ucsc'
tmp$NCBI_Build ='GRCh38'
tmp$NCBI_Build ='GRCh38'
tmp$Strand ='+'
tmp$Variant_Classification = tmp$effect
tail(sort(table(tmp$Variant_Classification )))
tmp$Tumor_Seq_Allele1 = tmp$Reference_Allele
tmp$Variant_Type = ifelse(
tmp$Reference_Allele %in% c('A','C','T','G') & tmp$Tumor_Seq_Allele2 %in% c('A','C','T','G'),
'SNP','INDEL'
)
table(tmp$Variant_Type )
tcga.brca = read.maf(maf = tmp,
vc_nonSyn=names(tail(sort(table(tmp$Variant_Classification )))))
oncoplot(maf = tcga.brca, top = 10) # 高频突变的前10个基因
出图如下所示:
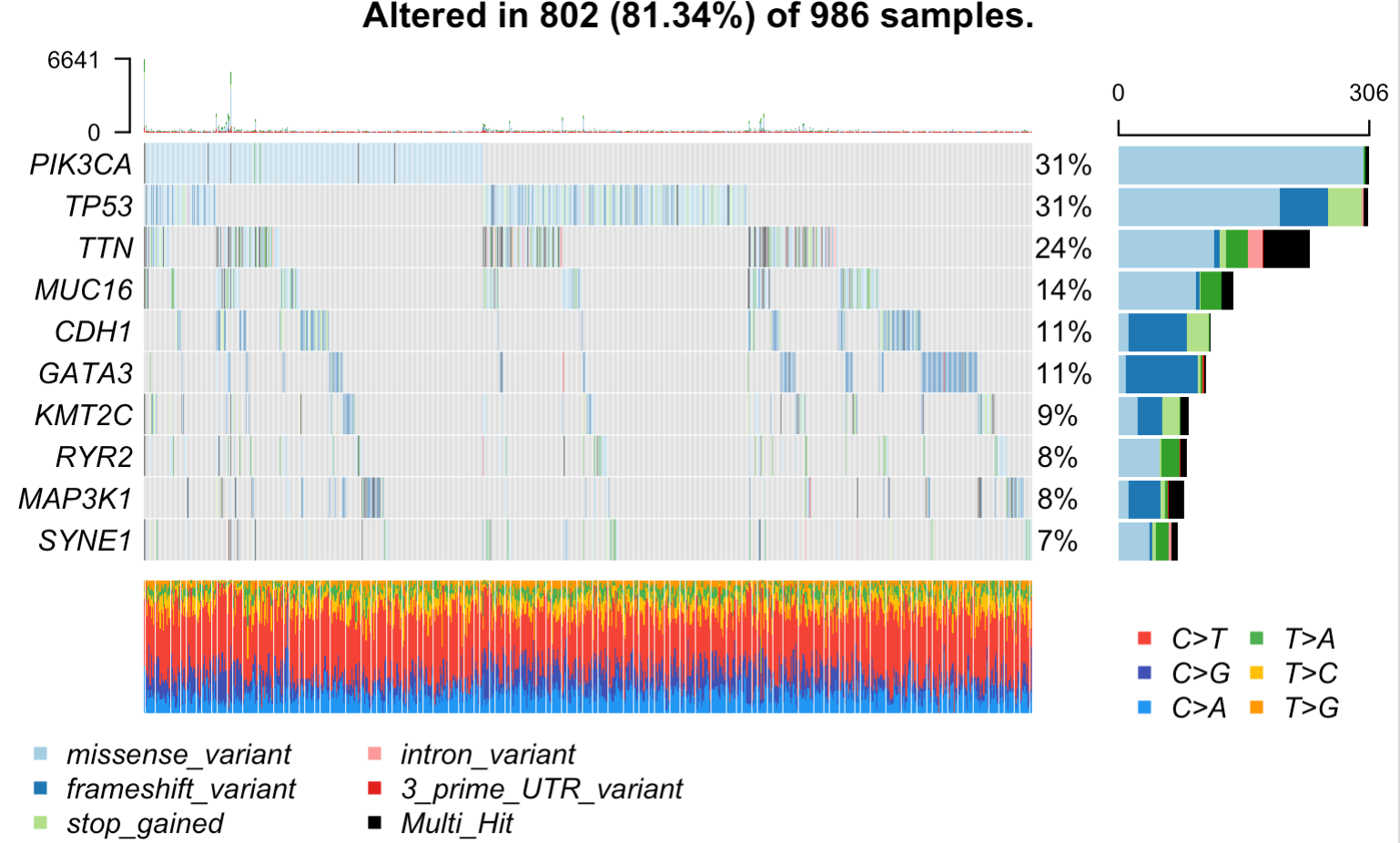
其实主要就是对maftools 这个包的read.maf函数的理解,以及对maf文件格式的理解。