单细胞转录组下游的降维聚类分群这样的基础操作相信大家应该是都没有问题了,参考前面的例子:人人都能学会的单细胞聚类分群注释 ,只需要你有一个基于基因的表达量矩阵的分析。
对转录组数据(包括单细胞转录组)以基因为单位定量是非常容易的,因为配套的gtf文件非常齐全啦。但是最近看到了一个有意思的文章,标题是:《Uncovering transcriptional dark matter via gene annotation independent single-cell RNA sequencing analysis》,链接是:https://www.nature.com/articles/s41467-021-22496-3 ,它里面是针对 transcriptionally active regions (TARs) 进行定量,拿到的表达量矩阵也是可以进行降维聚类分群,流程图如下所示:
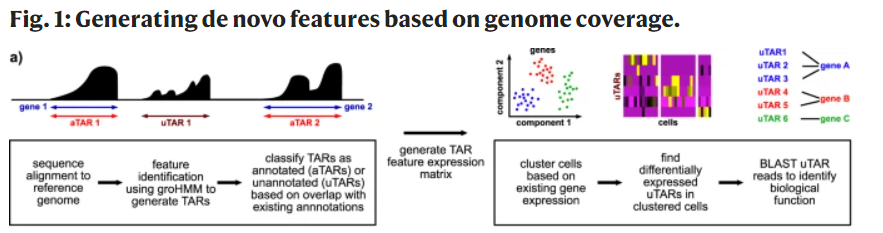
文章的 脚本代码和数据都是公开的:
- TAR-scRNA-seq tool is available at https://github.com/fw262/TAR-scRNA-seq with the identifier (https://doi.org/10.5281/zenodo.4567436).
这个groHMM的详细描述如下所示: - we used groHMM to scan the mapped read counts along the genome with window size 50 bp without overlap.
- The emission probabilities were modeled by a gamma distribution. The gamma distribution parameters and transition probabilities were learned using the Baum-Welch expectation maximization (EM) algorithm.
- We predicted the transcribed regions from both the sense and antisense strands. Predicted transcribed regions within 500 bp were merged using bedtools merge (parameters: -s -d 500).
- The coverage of each region was calculated using bedtools coverage (parameters: -s -counts -split).
- TARs identified using the groHMM algorithm were labeled as annotated TAR (aTAR) or unannotated TAR (uTAR) features based on their overlap with existing gene annotations.
这个 groHMM 其实是2015就发表了,文章是: groHMM: a computational tool for identifying unannotated and cell type-specific transcription units from global run-on sequencing data. BMC Bioinformatics 16, 222 (2015).
挺有意思的,其实单细胞转录组里面可以挖掘的点实在是太多了,另外一个类似的思路是分析聚腺苷酸化(APA)事件,我看到了:
- 李蔚教授首创性开发了一种可分析标准单细胞RNA测序数据(scRNA-seq) 多聚腺苷酸化(APA)事件的算法——scDaPars。将scDaPars分析真实乳腺癌Smart-seq2单细胞数据,发现与正常细胞相比,肿瘤细胞3’UTR显著缩短,并且依靠APA事件可以将肿瘤与非肿瘤细胞明显分开。
- 2021年8月10日,中国科学院上海营养与健康研究所杨力研究组在Genome Biology在线发表了题为 SCAPTURE: a deep learning-embedded pipeline that captures polyadenylation information from 3’ tag-based RNA-seq of single cells 的研究成果,通过建立一套新型的计算分析方法——SCAPTURE,利用3’ tag-based scRNA-seq开展单细胞水平的RNA多聚腺苷酸化及相关转录本水平基因表达的分析研究。 SCAPTURE软件已在GitHub发布:https://github.com/YangLab/SCAPTURE
其实也可以转为转录因子活性程度矩阵或者gsea等算法的基因集打分矩阵
转录因子活性程度的教程看:
- 2021年8月10日,中国科学院上海营养与健康研究所杨力研究组在Genome Biology在线发表了题为 SCAPTURE: a deep learning-embedded pipeline that captures polyadenylation information from 3’ tag-based RNA-seq of single cells 的研究成果,通过建立一套新型的计算分析方法——SCAPTURE,利用3’ tag-based scRNA-seq开展单细胞水平的RNA多聚腺苷酸化及相关转录本水平基因表达的分析研究。 SCAPTURE软件已在GitHub发布:https://github.com/YangLab/SCAPTURE
- 张泽民团队的单细胞研究把T细胞分的如此清楚
- 细胞通讯分析的背景知识
- 构建单细胞亚群网络(类似于细胞通讯分析)
- 细胞通讯分析结果的解读
- SCENIC转录因子分析结果的解读
- 人人都能学会的单细胞聚类分群注释
- 对单细胞表达矩阵做gsea分析
- 单细胞转录因子分析之SCENIC流程
gsea等算法的基因集打分矩阵看:借鉴escape包的一些可视化GSVA或者ssGSEA结果矩阵的方法 以及 对单细胞表达矩阵做gsea分析,代码实现环节大同小异。