我们以前分享过:GEO数据挖掘技术可以应用到表达芯片也可以是转录组测序,但是呢,RNA-seq测序数据并不会把其表达矩阵存储在Series Matrix File(s) 里面,所以 你使用我的标准代码:
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)#在调用as.data.frame的时,将stringsAsFactors设置为FALSE可以避免character类型自动转化为factor类型
# 注意查看下载文件的大小,检查数据
f='GSE103611_eSet.Rdata'
# https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE103611
library(GEOquery)
# 这个包需要注意两个配置,一般来说自动化的配置是足够的。
#Setting options('download.file.method.GEOquery'='auto')
#Setting options('GEOquery.inmemory.gpl'=FALSE)
if(!file.exists(f)){
gset <- getGEO('GSE103611', destdir=".",
AnnotGPL = F, ## 注释文件
getGPL = F) ## 平台文件
save(gset,file=f) ## 保存到本地
}
load('GSE103611_eSet.Rdata') ## 载入数据
class(gset) #查看数据类型
length(gset) #
class(gset[[1]])
gset
# assayData: 352859 features, 48 samples
是可以拿到表达量矩阵,因为https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE103611 里面的是 Affymetrix Human Gene 2.0 ST Array [probe set (exon) version] 的表达量芯片。
但是对 对 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE106292 上面的代码就拿不到表达矩阵 ,因为 是转录组测序数据。其 提供的表达量矩阵是 GSE106292_Human_Matrix_final.xlsx 这个 4.6 Mb,就很麻烦,因为它并不是标准的counts矩阵,不一定适合于 edgeR、DEseq2这样的包!
但是最近给学徒安排了一个类似的仅仅是提供了rpkm这样的归一化并且带小数点的转录组表达量矩阵项目做差异分析,发现了一个骚操作,我也是醉了。如下所示,rpkm矩阵仍然是可以把肿瘤和正常组织泾渭分明的区分开来。
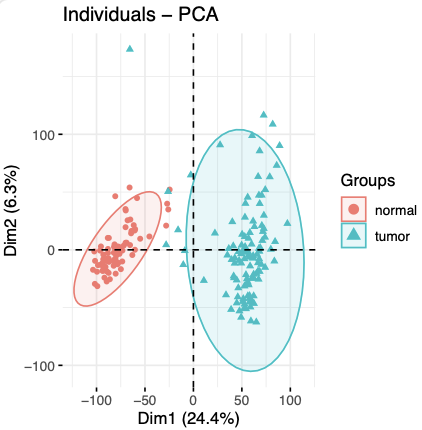
学徒做了差异分析,然后 上下调各自选取100个差异基因,热图可视化如下所示:
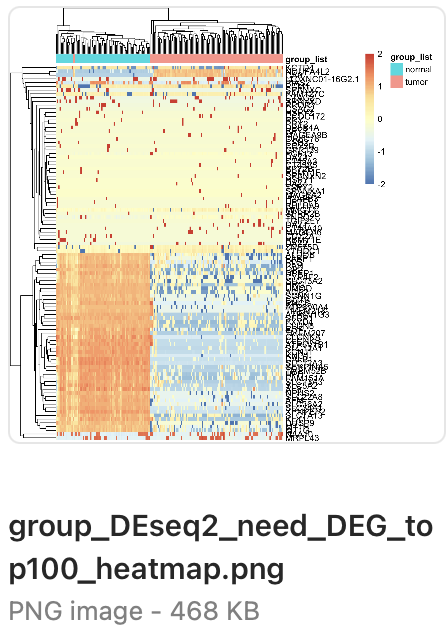
看起来分析的有模有样!简单了问了,才知道,学徒仅仅是根据我的表达芯片的公共数据库挖掘系列推文 :
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
代码如下所示:
> mat[1:4,1:3] # rpkm 格式的表达量矩阵
C3L-00418_Solid Tissue Normal C3L-00004_Primary Tumor C3N-00852_Primary Tumor
OR4F5 0.0000000 0.0000000 0.0000000
SAMD11 4.0715358 0.3719551 0.6190211
KLHL17 2.3878834 2.1623200 1.6117428
PLEKHN1 0.3629224 0.4835416 1.5632905
> exprSet=log2(mat+1)
> # 为了使用edgeR、DEseq2
> exprSet <- ceiling(exprSet)
> exprSet[1:4,1:3]
C3L-00418_Solid Tissue Normal C3L-00004_Primary Tumor C3N-00852_Primary Tumor
OR4F5 0 0 0
SAMD11 3 1 1
KLHL17 2 2 2
PLEKHN1 1 1 2
也就是说,学徒仅仅是把下载好了的 rpkm 格式的表达量矩阵进行了log2和取整的操作, 然后就假装它是一个 整数矩阵,去走 edgeR、DEseq2这样的专门为转录组测序counts矩阵开发的差异分析流程!
我让学徒重新走转录组测序数据分析流程,拿到真正的counts矩阵,再做差异分析,然后比较两次差异分析结果。
#画九宫格就是上下调基因画在一起,这样用logFC=1画
#~~~~~~数据进行比较~~~~~~~
# deg_gdc 是DEseq2包对 counts矩阵的差异分析结果
# deg_cptac 是DEseq2包对 rpkm 矩阵的差异分析结果
int_gene=intersect(deg_gdc$V1,deg_cptac$V1)
head(int_gene)
length(int_gene)# 17568
comp=cbind(deg_gdc[int_gene,],
deg_cptac[int_gene,])
head(comp)
dim(comp)
colnames(comp)
#~~~~~看上下调基因的交集~~~~~
table(comp[,c(8,16)])
# change.1
# change DOWN stable UP
# DOWN 611 1568 0
# stable 11 12647 1
# UP 0 2387 343
#~~~~~初步画图~~~~~
plot(comp[,c(3,11)])
dev.off()
#~~~~进阶画图~~~~
comp_logFC <- comp[,c(3,8,11,16)]
head(comp_logFC)
logFC_t = 1
#P.Value_t = 0.05
p <- ggplot(comp_logFC, aes(x=comp_logFC$log2FoldChange, y=comp_logFC$log2FoldChange.1))+
geom_point()+
labs(x = "GDC",
y = "CPTAC")+
scale_color_manual(values=c("blue", "grey","red"))+
geom_vline(xintercept = c(-logFC_t,logFC_t),lty=4,col="#ec183b",lwd=0.8) +
geom_hline(yintercept = c(-logFC_t,logFC_t),lty=4,col="#18a5ec",lwd=0.8) +
xlim(-5,5)+
ylim(-3,3)+
theme_ggstatsplot()+
theme(panel.grid.minor = element_blank(),
panel.grid.major = element_blank())
p
ggsave(filename = 'sudoku.png',width = 10,height = 8)
dev.off()
有意思的地方出现了,
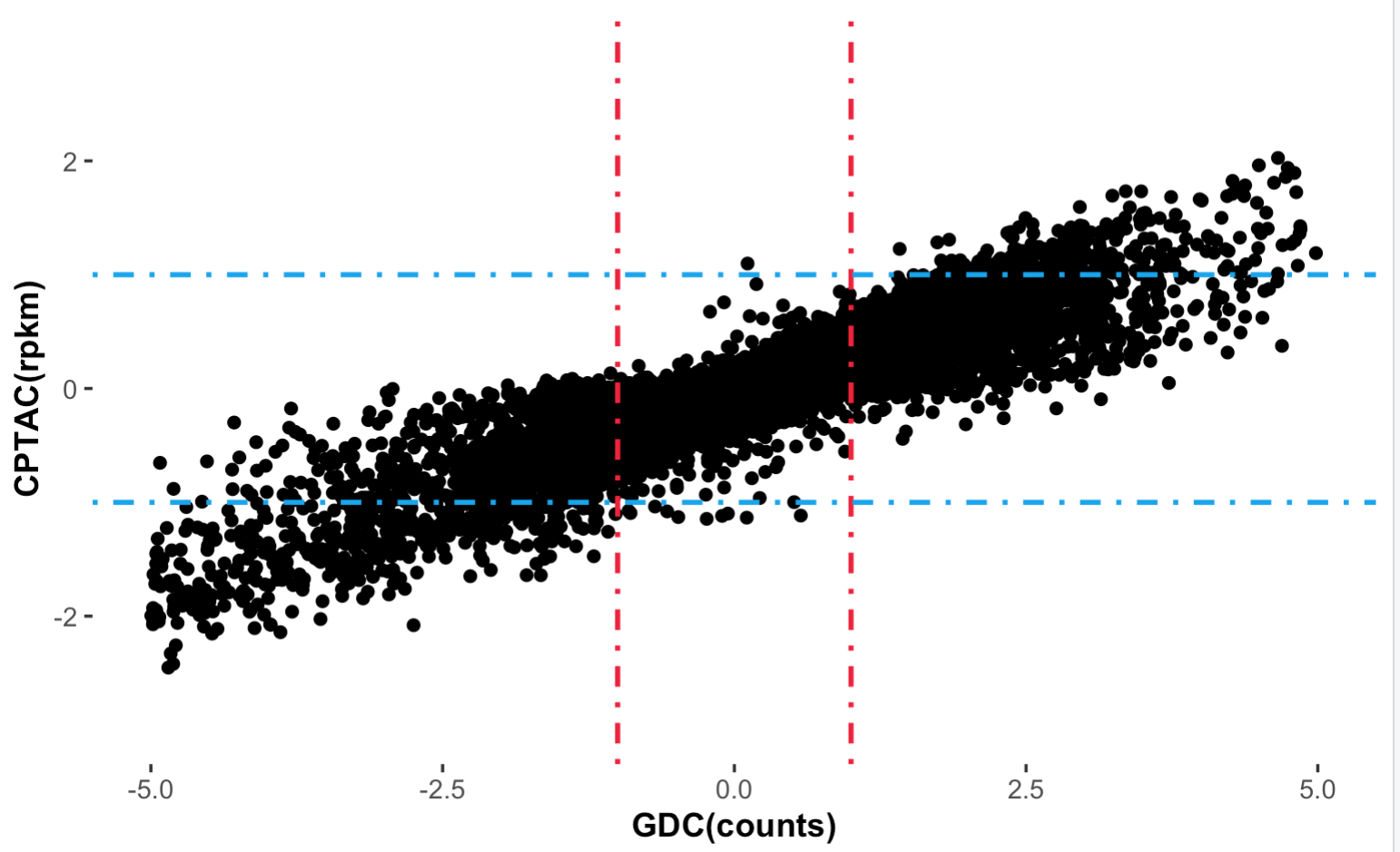
两次差异分析的结果居然是出奇的吻合,至少是从变化倍数的角度来看!
有意思