在前面的教程:居然有如此多种癌症(是时候开启pan-cancer数据挖掘模式),我们把全部的TCGA的33种癌症的表达量矩阵区拆分成为蛋白编码基因和非编码基因这两个不同的表达量矩阵,并且保存成为了rdata文件。
在不同癌症里面,蛋白质编码相关基因数量一直在一万八附近,而非编码基因数量跨度比较大,从一万二到两万七不等。因为非编码基因的组织病人特异性都很强。
接下来,我们整合全部的33种癌症的仅仅是蛋白质编码基因的表达量矩阵:
首先是 合并全部癌症表达量矩阵,自己走PCA,tSNE,DBSCAN , 代码如下:
fs=list.files('Rdata/',pattern = 'htseq_counts')
fs
# 首先加载每个癌症的蛋白编码基因表达量矩阵
# 成为一个 list
dat_list = lapply(fs, function(x){
# x=fs[1]
pro=gsub('.htseq_counts..Rdata','',x)
print(pro)
load(file = file.path('Rdata/',x))
dat=log2(edgeR::cpm(pd_mat)+1)
return(dat)
})
tmp=table(unlist(lapply(dat_list, rownames)))
cg=names(tmp)[tmp==33]
length(cg)
# 在33个癌症都存在的蛋白编码基因是一万六千个
all_tcga_matrix=do.call(cbind,
lapply(dat_list, function(x){
x[cg,]
}))
dim(all_tcga_matrix)
group=rep(gsub('TCGA-','',gsub('.htseq_counts..Rdata','',fs)),
unlist(lapply(dat_list, ncol)))
table(group)
各个癌症的样品数量如下所示:
> table(group)
group
ACC BLCA BRCA CESC CHOL COAD DLBC ESCA GBM
79 430 1217 309 45 512 48 173 173
HNSC KICH KIRC KIRP LAML LGG LIHC LUAD LUSC
546 89 607 321 151 529 424 585 550
MESO OV PAAD PCPG PRAD READ SARC SKCM STAD
86 379 182 186 551 177 265 472 407
TGCT THCA THYM UCEC UCS UVM
156 568 121 583 56 80
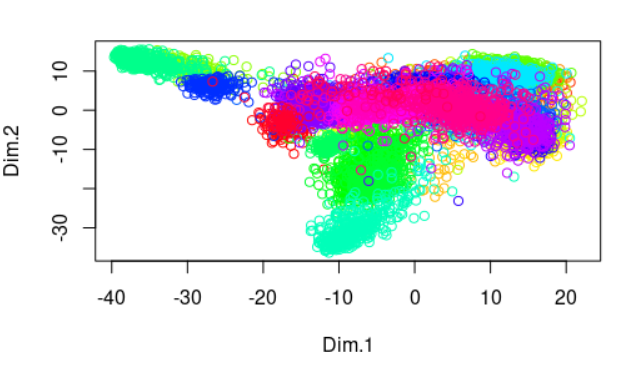
首先看最基础的pca
# 为了加快运算速度,我们取top1000基因即可,按照sd派系
cg=names(tail(sort(apply(all_tcga_matrix,1,sd)),1000))
# 首先查看pca情况
library("FactoMineR")
library("factoextra")
dat.pca <- PCA(t(all_tcga_matrix[cg,]), graph = FALSE)
fviz_pca_ind(dat.pca,
repel =T,
geom.ind = "point",
col.ind = group,
addEllipses = TRUE,
legend.title = "Groups"
)
plot(dat.pca$ind$coord[,1:2],
col=rainbow(33)[as.numeric(as.factor(group))] )
你会分析33种癌症,区分度很差:
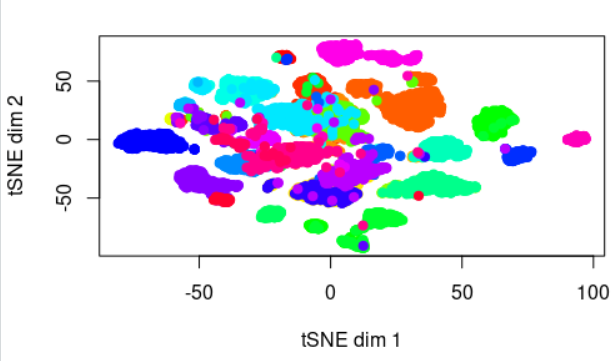
接着看tSNE
library(Rtsne)
tsne_out <-Rtsne(t(all_tcga_matrix[cg,]),
initial_dims=30,verbose=FALSE,
check_duplicates=FALSE,
perplexity=27, dims=2,max_iter=5000)
plot(tsne_out$Y,
pch=19, xlab="tSNE dim 1", ylab="tSNE dim 2",
col=rainbow(33)[as.numeric(as.factor(group))])
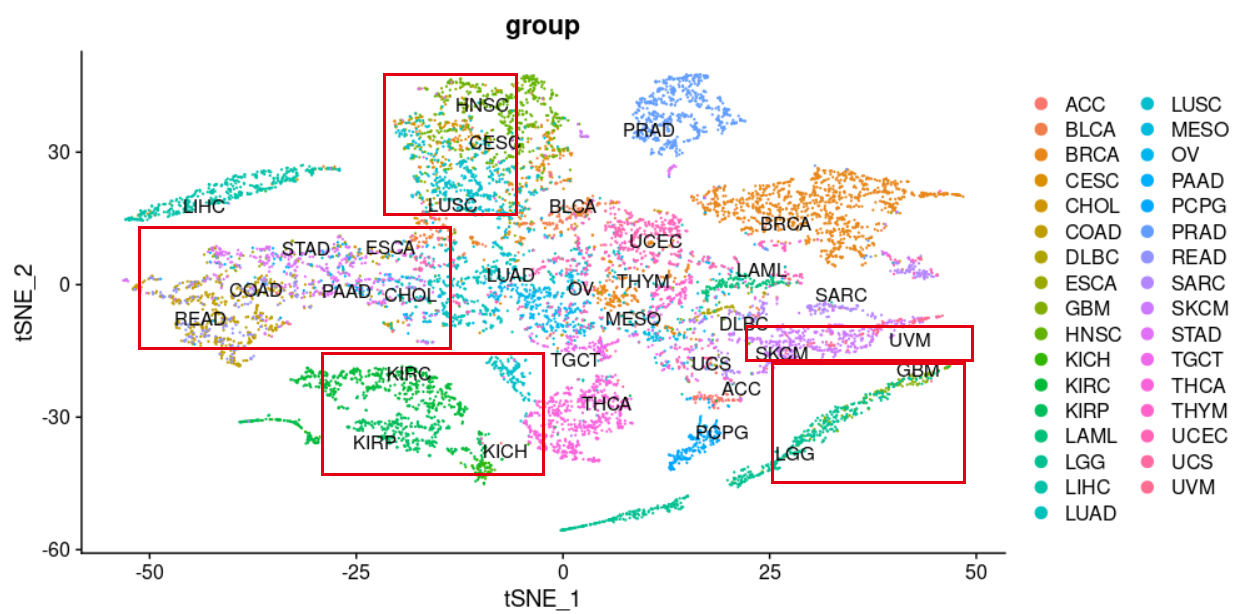
顿时不同癌症的区分度就好很多啦:
可以比较kmeans和dbscan的差异
有了上面的tSNE坐标,可以看到不同的癌症有比较好的区分度,但是里面呢仍然是会有混淆,我们可以看看自己根据tSNE坐标的分群结果,跟病人本身的癌症属性是否一致!
library(gplots)
kmeans_cluster_tsne=kmeans(tsne_out$Y,centers = 33)$clust
library(dbscan)
dbscan_cluster_tsne=dbscan(tsne_out$Y,eps=3.1)$cluster
balloonplot(table(dbscan_cluster_tsne,
kmeans_cluster_tsne))
balloonplot(table(dbscan_cluster_tsne,
group))
plot(tsne_out$Y,col=kmeans_cluster_tsne,
pch=19, xlab="tSNE dim 1", ylab="tSNE dim 2")
plot(tsne_out$Y,col=dbscan_cluster_tsne,
pch=19, xlab="tSNE dim 1", ylab="tSNE dim 2")
library(RColorBrewer)
plot(tsne_out$Y,col=kmeans_cluster_tsne,
pch=19, xlab="tSNE dim 1", ylab="tSNE dim 2")
plot(tsne_out$Y,col=dbscan_cluster_tsne,
pch=19, xlab="tSNE dim 1", ylab="tSNE dim 2")
可以看到还是蛮有意思的:
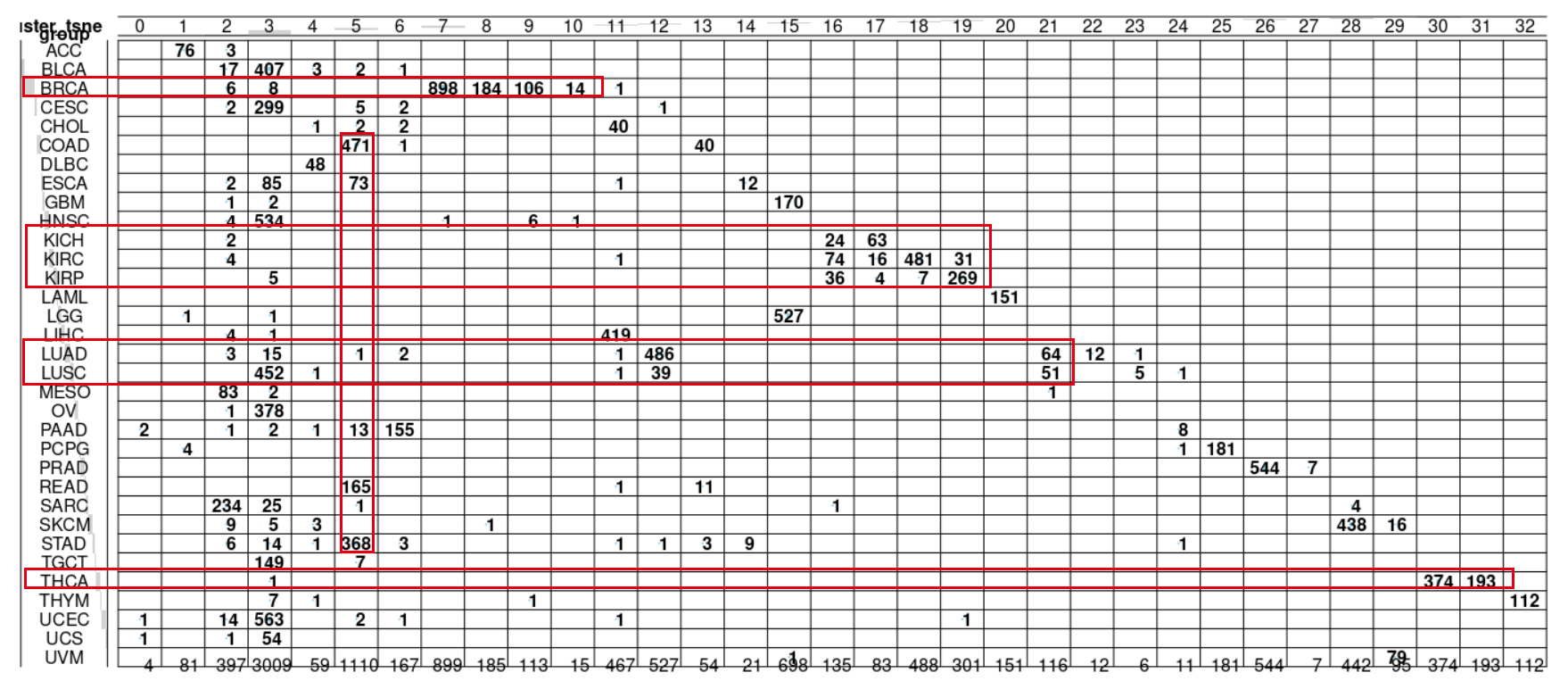
其中很明显乳腺癌区分成为了不同亚群,应该是对应着不同的分子分型,尤其是三阴性或者说basal-like的,跟乳腺癌主群区分很开。
而KICH,KIRC,KIRP 这3种肾癌,互相之间纠缠不清。
LUAD和LUSC两个肺癌也是有部分混淆。
其中COAD,READ,STAD主要的消化道癌症被聚类在了一起,也许是因为他们的性质过于类似吧。
让我意外的是THCA居然也被拆分成为了两个主要的小群,这个是我的知识盲区了。
我们这个小单元的标题是比较kmeans和dbscan的分群差异,那么,两个算法分群差异到底一致性如何呢?同样是看图:
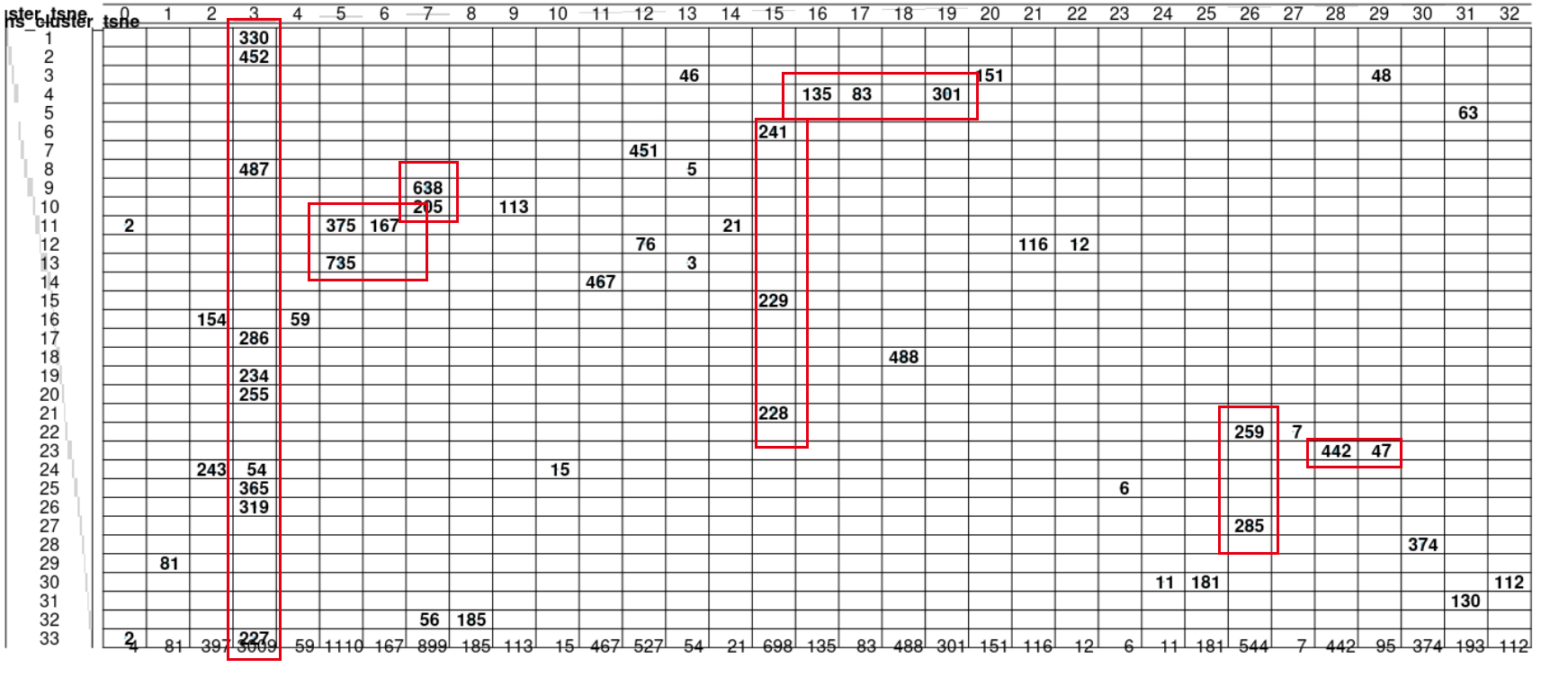
差异肯定是有,但是如何量化, 这是一个好问题。
另外, 我们都提到了大样本量多分组的表达量矩阵,其实可以干脆使用单细胞流程啦,下期我们再见,使用seurat流程重新分析,可以出如下所示图表: