我们做肿瘤研究的单细胞数据,一般来说会选择初步很粗狂的定义大的细胞亚群,比如我常用的 第一次分群是通用规则是:
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibo or CD31+,PECAM1,endo)
然后绝大部分文章都是抓住免疫细胞亚群进行细分,包括淋巴系(T,B,NK细胞)和髓系(单核,树突,巨噬,粒细胞)的两大类作为第二次细分亚群。 说起来很简单,但是实际上每次做到单细胞数据集的细分亚群就非常的头疼,尤其是myeloid的髓系,(单核,树突,巨噬,粒细胞)有时候根本就分不清楚,而且分完之后仍然是可以继续细分。
最近我分享了一个单细胞转录组文献,《Single-cell transcriptomics reveals regulators underlying immune cell diversity and immune subtypes associated with prognosis in nasopharyngeal carcinoma》,视频可以在b站看到;
我尝试对它这个数据集进行数据分析图表复现,比如单独把髓系拿出来进行重新降维聚类分群,然后可视化如下所示:
load(file = 'sce_recluster.Rdata')
p1=DimPlot(sce,reduction = "umap",label=T
,group.by = 'Cell_type')
p2=DimPlot(sce,reduction = "umap",label=T
,group.by = 'orig.ident')
table(sce$orig.ident,sce$seurat_clusters)
p3=DimPlot(sce,reduction = "umap",label=T)
library(patchwork)
p1/p2/p3
可以看到pDC这个细胞亚群,由 4和8群组成,而且包含多个病人!
但是p07这个病人就非常的诡异,这个样品里面的多种髓系细胞居然是与其它病人样品的髓系细胞距离超级远!
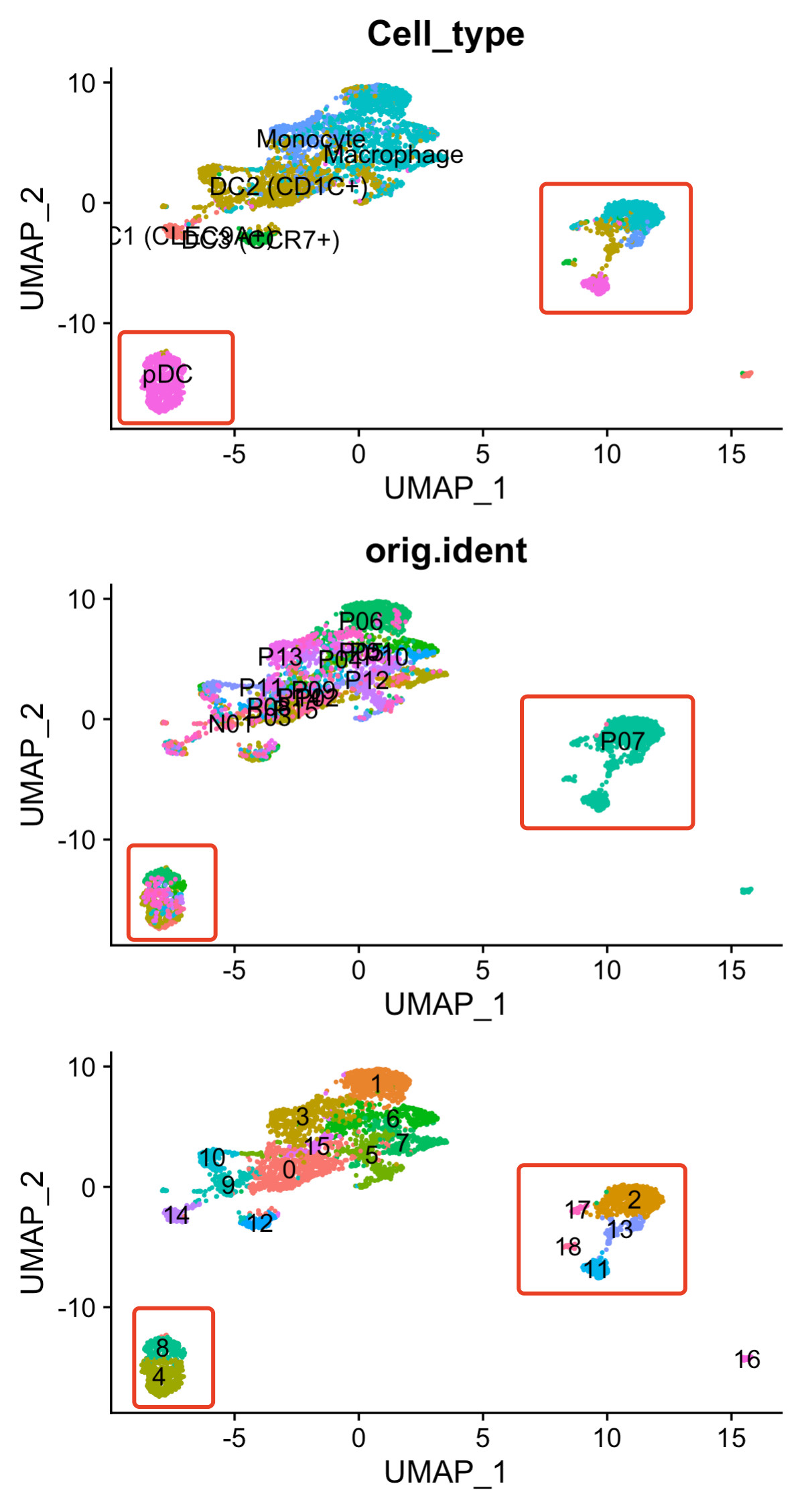
但是如果你harmony处理一下,然后再降维聚类分群,代码如下所示:
load(file = 'main_sce_recluster.Rdata')
sce.all.filt=sce
library(harmony)
sce.all.int <- RunHarmony(sce.all.filt,
c( "orig.ident" ))
names(sce.all.int@reductions)
harmony_embeddings <- Embeddings(sce.all.int, 'harmony')
harmony_embeddings[1:5, 1:5]
sce.all.int=RunTSNE(sce.all.int,reduction = "harmony", dims = 1:30)
sce.all.int=RunUMAP(sce.all.int,reduction = "harmony",dims = 1:10)
sce=sce.all.int
sce <- FindNeighbors(sce, reduction = "harmony",dims = 1:15)
sce <- FindClusters(sce, resolution = 0.8)
load(file = 'after_harmony/main_sce_recluster.Rdata')
p1=DimPlot(sce,reduction = "umap",label=T
,group.by = 'Cell_type')
p2=DimPlot(sce,reduction = "umap",label=T
,group.by = 'orig.ident')
table(sce$orig.ident,sce$seurat_clusters)
p3=DimPlot(sce,reduction = "umap",label=T)
library(patchwork)
p1/p2/p3
出图如下:
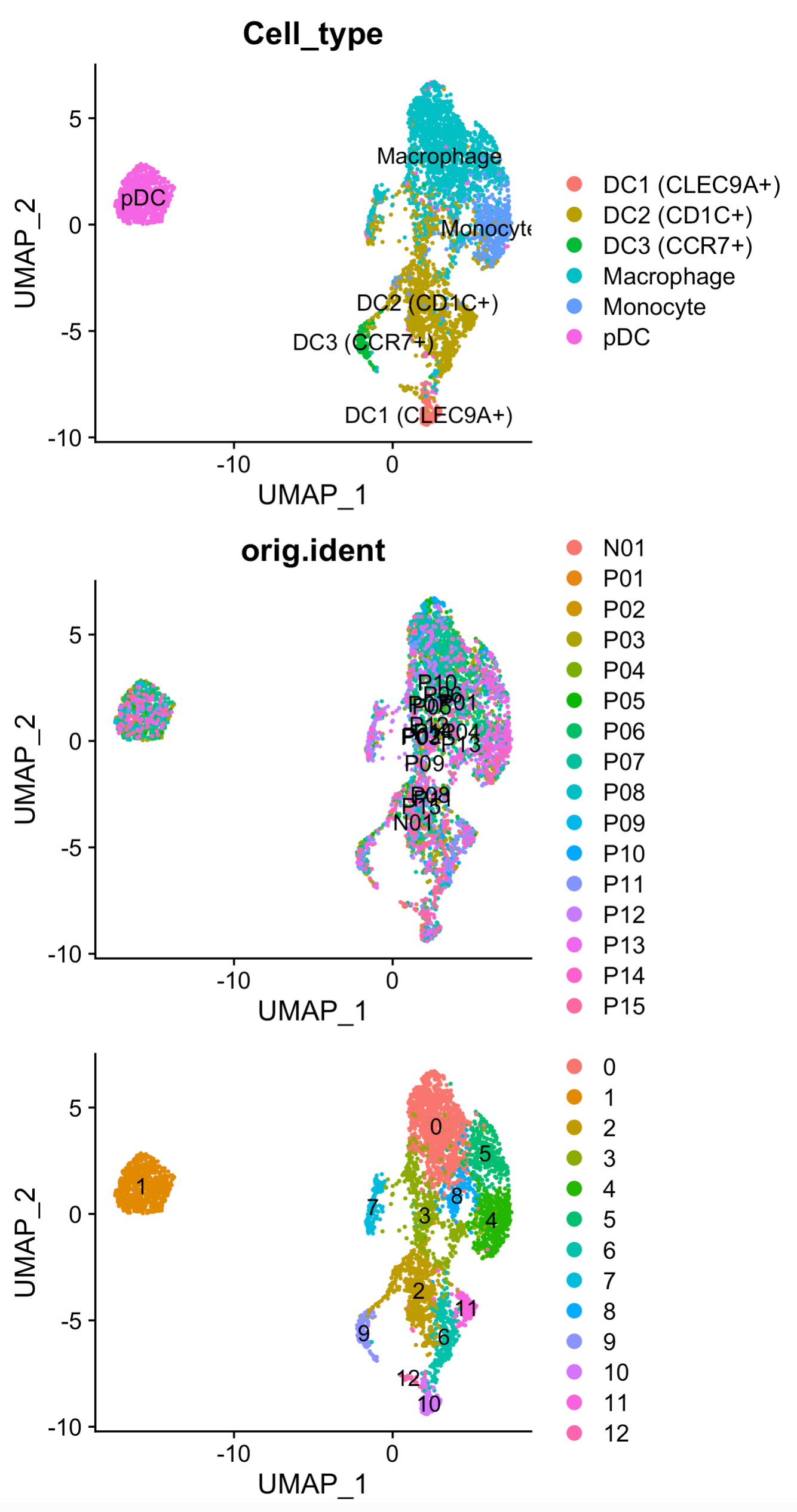
可以看到这个时候的p07这个病人不会在独立成为一个亚群啦,而且呢,pDC这个细胞亚群也比较纯粹一点了。
真棒啊!
如果你对单细胞数据分析还没有基础认知,可以看基础10讲: