前面我们分享过:学徒数据挖掘之谁说生存分析一定要按照表达量中位值或者平均值分组呢?,可以很容易对一个基因,根据表达量分组,然后进行生存分析,判断它是风险因子或者是保护因子,当然了前提是具有统计学显著性啦。
然后很多粉丝留言说,如果并不是按照表达量中位值或者平均值分组,而是取巧使用了surv_cutpoint这样的函数,得到的结果并不好解释,认为这样的的数据处理方式简直是黑白颠倒!
我看到了这样的留言,确实思考了一下,使用了surv_cutpoint这样的函数是仅仅是改变了统计学显著性指标呢?还是说,确实有可能造成黑白颠倒呢?风险因子会变成保护因子???
布置它为学徒作业吧
前面自己把表达量矩阵和临床信息准备好,得到 exprSet这个表达量矩阵,以及 meta 这个临床信息,然后就可以使用了surv_cutpoint这样的函数对指定基因做生存分析啦,代码如下所示:
library(survival)
library(survminer)
gs=c('BCL2','BCL2L1','MCL1')
splots <- lapply(gs, function(g){
meta$gene = t(exprSet[g,])
sur.cut <- surv_cutpoint(meta, time= 'time',
event = 'event' ,
variables = 'gene' )
sur.cat <- surv_categorize(sur.cut)
sfit1=survfit(Surv(time, event)~gene, data=sur.cat)
ggsurvplot(sfit1,pval =TRUE, data = meta)
})
arrange_ggsurvplots(splots, print = TRUE,
ncol = 2, nrow = 2, risk.table.height = 0.4)
如下所示;
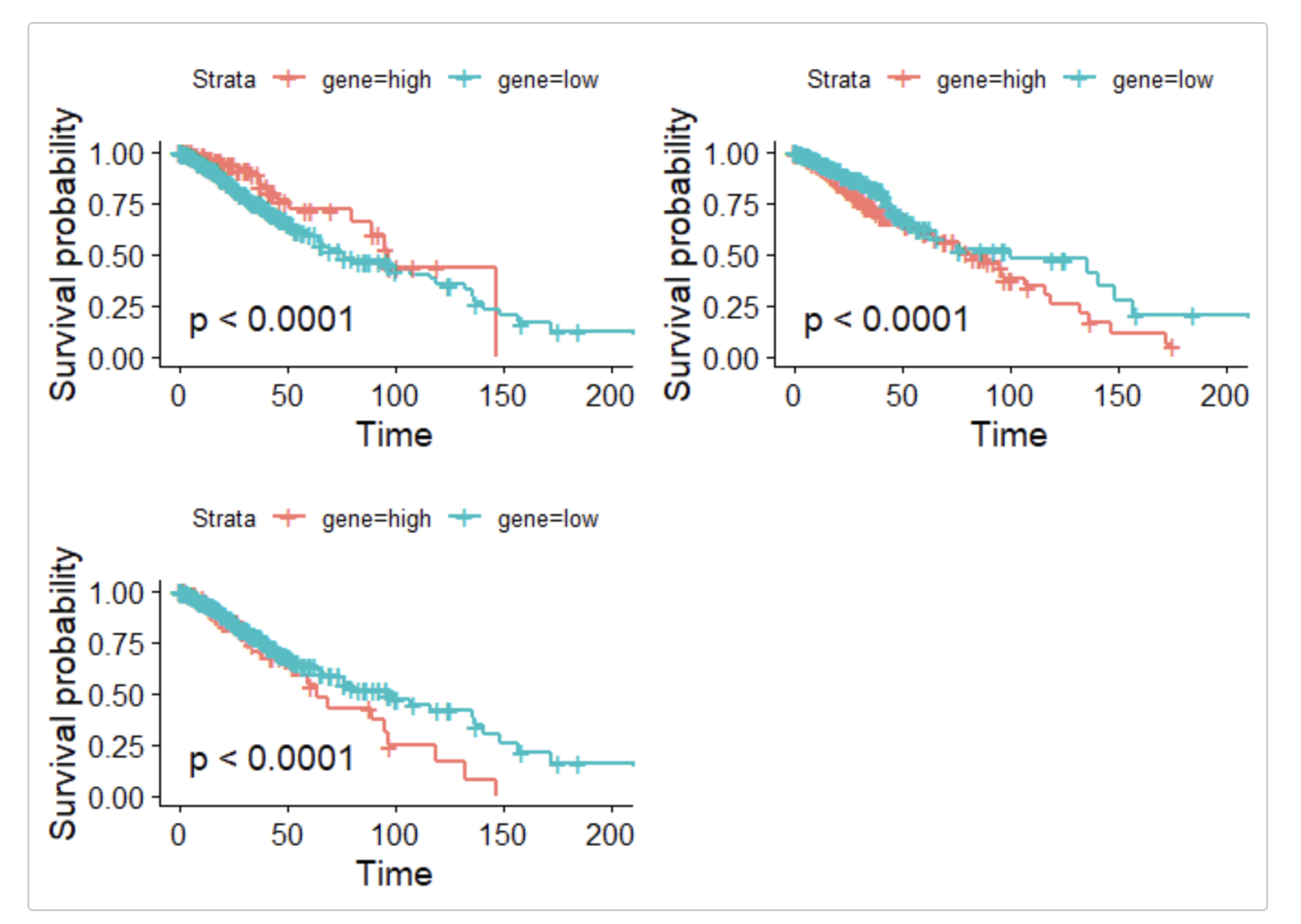
这个 surv_cutpoint 函数确实效果给力,基本上没有它搞不定的统计学显著性。
表达量矩阵和临床信息的准备过程
需要自己去tcga所在的ucsc的xena网站 ,去下载 TCGA.LGG.sampleMap_HiSeqV2.gz 文件以及 TCGA.LGG.sampleMap_LGG_clinicalMatrix 文件,然后下面的代码就可以复制粘贴直接运行啦!
1.准备数据
rm(list = ls())
library(stringr)
exp = read.table('../TCGA.LGG.sampleMap_HiSeqV2.gz',header = T,row.names = 1,check.names = F)
exp = as.matrix(exp)
clinical = data.table::fread('../TCGA.LGG.sampleMap_LGG_clinicalMatrix')
dim(exp)
#> [1] 20530 530
clinical[1:4,1:4]
#> sampleID _INTEGRATION _PATIENT _cohort
#> 1: TCGA-CS-4938-01 TCGA-CS-4938-01 TCGA-CS-4938 TCGA Lower Grade Glioma (LGG)
#> 2: TCGA-CS-4941-01 TCGA-CS-4941-01 TCGA-CS-4941 TCGA Lower Grade Glioma (LGG)
#> 3: TCGA-CS-4942-01 TCGA-CS-4942-01 TCGA-CS-4942 TCGA Lower Grade Glioma (LGG)
#> 4: TCGA-CS-4943-01 TCGA-CS-4943-01 TCGA-CS-4943 TCGA Lower Grade Glioma (LGG)
2.数据过滤
#过滤一下
dim(exp)
#> [1] 20530 530
table(str_sub(colnames(exp),14,15))
#>
#> 01 02
#> 516 14
# 01肿瘤,02复发,11正常
group_list = ifelse(as.numeric(str_sub(colnames(exp),14,15)) < 10,'tumor','normal')
group_list = factor(group_list,levels = c("normal","tumor"))
table(group_list)
#> group_list
#> normal tumor
#> 0 530
#这里是根据较小样本(normal)取值,当然可以有别的取值
exp = exp[apply(exp, 1, function(x) sum(x > 1) > 0), ]
dim(exp)
#> [1] 19713 530
exp[1:4,1:4]
#> TCGA-E1-5319-01 TCGA-HT-7693-01 TCGA-CS-6665-01 TCGA-S9-A7J2-01
#> ARHGEF10L 10.1208 11.8084 10.1872 10.9576
#> HIF3A 10.4506 10.5173 3.6466 4.8099
#> RNF17 0.0000 0.0000 0.0000 0.4657
#> RNF10 11.2171 11.8974 11.5369 11.2675
#save(exp,group_list,clinical,file = '../data/01TCGA-LGG.Rdata')
3.生存分析前准备
这里可以选取自己需要的列,
### 1.简化临床信息,选出要用的列
tmp = data.frame(colnames(clinical))
#colnames(clinical)
#write.csv(clinical,file = 'clinical.csv')
clinical = clinical[,c(
'bcr_patient_barcode',
'vital_status',
"days_to_last_followup" ,
'days_to_death',
'days_to_birth'
)]
dim(clinical)
#> [1] 530 5
#rownames(clinical) <- NULL
clinical = clinical[!duplicated(clinical$bcr_patient_barcode),]
rownames(clinical) <- clinical$bcr_patient_barcode
clinical[1:4,1:4]
#> bcr_patient_barcode vital_status days_to_last_followup days_to_death
#> 1: TCGA-CS-4938 LIVING 3574 NA
#> 2: TCGA-CS-4941 DECEASED NA 234
#> 3: TCGA-CS-4942 DECEASED NA 1335
#> 4: TCGA-CS-4943 DECEASED 481 1106
meta = clinical
exprSet=exp[,group_list=='tumor']
table(group_list)
#> group_list
#> normal tumor
#> 0 530
exprSet = log2(edgeR::cpm(exprSet)+1)
#简化meta的列名
colnames(meta)=c('ID','event','last_followup','death','age')
#空着的值改为NA
meta[meta==""]=NA
### 2.实现表达矩阵与临床信息的匹配
# 以病人为中心,表达矩阵按病人ID去重复
k = !duplicated(str_sub(colnames(exprSet),1,12));table(k)
#> k
#> FALSE TRUE
#> 14 516
exprSet = exprSet[,k]
exprSet = as.data.frame(exprSet)
#调整meta的ID顺序与exprSet列名一致
exprSet = exprSet[,str_sub(colnames(exprSet),1,12) %in% meta$ID]
meta=meta[match(str_sub(colnames(exprSet),1,12),meta$ID),]
identical(meta$ID,str_sub(colnames(exprSet),1,12))
#> [1] TRUE
4.整理生存分析输入数据
#1.1计算生存时间(月)
meta$time = ifelse(meta$event=="LIVING",
meta$last_followup,
meta$death)
meta$time = as.numeric(meta$time)/30
#1.2 根据生死定义event,活着是0,死的是1
meta$event=ifelse(meta$event=='LIVING',
0,
1)
table(meta$event)
#>
#> 0 1
#> 389 126
#1.3 年龄和年龄分组
meta$age=ceiling(abs(as.numeric(meta$age))/365)
meta$age_group=ifelse(meta$age>median(meta$age,na.rm = T),'older','younger')
table(meta$age_group)
#>
#> older younger
#> 246 268
#save(meta,exprSet,file = "../data/TCGA-LGG_sur_model.Rdata")
得到 exprSet这个表达量矩阵,以及 meta 这个临床信息,后续的全部生存分析相关,都是依赖于它哦!
作业
对全部的基因,首先使用表达量中位值进行分组后,批量进行 cox 分析,拿到HR值和P值,输出成为表格。
然后对基因根据surv_cutpoint函数进行分组后,再批量cox分析,拿到HR值和P值,输出成为表格。
比较两个表格,看看是否有基因的HR值的方向冲突了,还是说,仅仅是统计学指标的改变。