前面我们已经是完整的展示了使用R语言的scenic做转录因子分析的流程,但是在公开课演示100个细胞走这个流程,发现居然是耗时20min,实在是不能忍。哪怕是再不喜欢python,也得学一学pyscenic了!
毕竟在文章《A scalable SCENIC workflow for single-cell gene regulatory network analysis》,有这个时间消耗对比,不服不行!
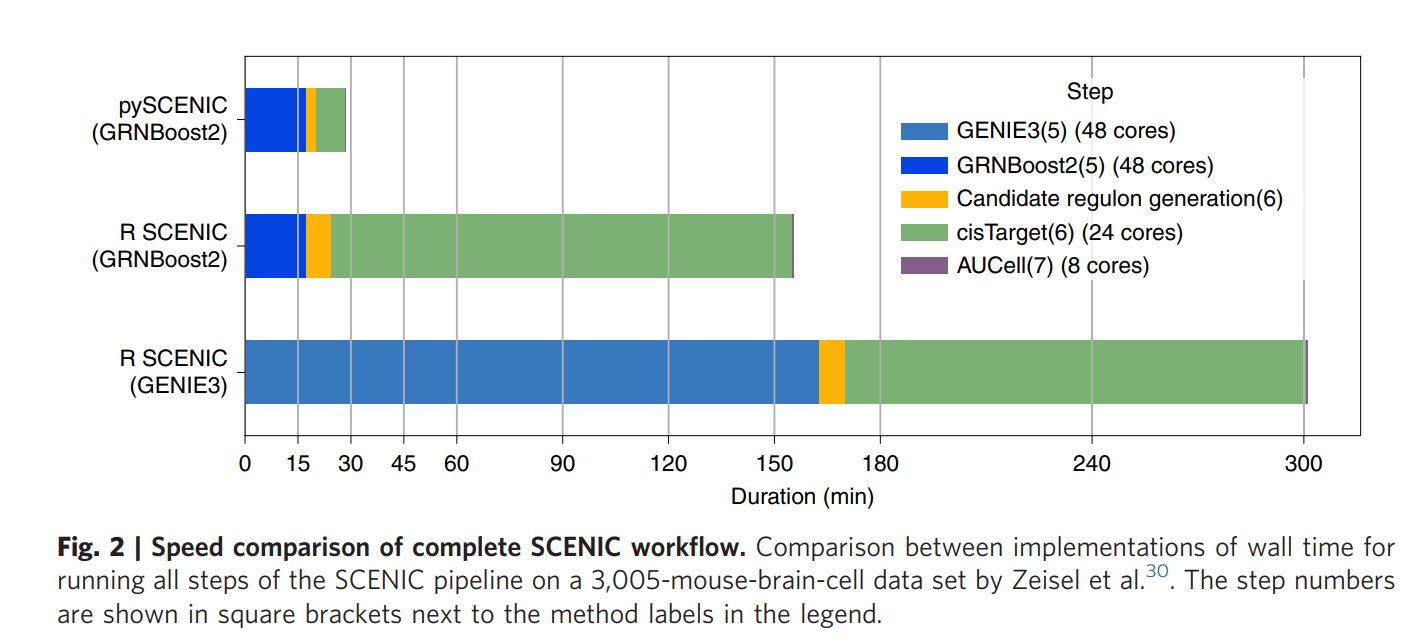
一般来说pyscenic是在Linux相关服务器上面操作,所以我也是在自己的ubuntu操作系统演示:
安装自己的conda,每个用户独立操作
安装方法代码如下:
# 首先下载文件,20M/S的话需要几秒钟即可
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
# 接下来使用bash命令来运行我们下载的文件,记得是一路yes下去
bash Miniconda3-latest-Linux-x86_64.sh
# 安装成功后需要更新系统环境变量文件
source ~/.bashrc
安装好conda后需要设置镜像。
conda config --add channels r
conda config --add channels conda-forge
conda config --add channels bioconda
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/cloud/bioconda/
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/pkgs/main/
conda config --set show_channel_urls yes
使用conda安装pyscenic的一些依赖
极端情况下,建议每个软件都可以独立给一个环境,虽然是这样会导致很多冗余的软件,不过,确实很方便。
# 需要一些依赖,尤其是这个python 版本
conda create -n pyscenic python=3.7
conda activate pyscenic
conda install -y numpy
conda install -y -c anaconda cytoolz
conda install -y scanpy
pip install pyscenic
有时候会遇到网络报错,换个时间点就好了
pip._vendor.urllib3.exceptions.ReadTimeoutError: HTTPSConnectionPool(host='files.pythonhosted.org', port=443): Read timed out.
然后,有意思的地方就来了,我测试了五台服务器,发现并不是所有的服务器都可以成功安装它!后面我们会讲解一个docker的解决方案,假如你确实只有的conda无法安装,也可以等明天的教程哈!
提取单细胞表达量矩阵csv忘记并且导入Linux服务器
首先我们对文章《Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma》的单细胞转录组数据进行降维聚类分群,然后提取fibo这个子亚群,然后再随机挑取1000个fibo细胞,这样的表达量矩阵进行后续分析。
在seurat里面将矩阵筛选,然后输出成csv,再用python读入,然后打包成 loom
#注意矩阵一定要转置,不然会报错
write.csv(t(as.matrix(fibo@assays$RNA@counts)),file = "fibo_1000.csv")
自己制作一个python 脚本,可以命名为 change.py , 内容如下所示:
import os, sys
os.getcwd()
os.listdir(os.getcwd())
import loompy as lp;
import numpy as np;
import scanpy as sc;
x=sc.read_csv("fibo_1000.csv");
row_attrs = {"Gene": np.array(x.var_names),};
col_attrs = {"CellID": np.array(x.obs_names)};
lp.create("sample.loom",x.X.transpose(),row_attrs,col_attrs);
然后运行这个脚本即可, python change.py ,这个命令就会读取当前文件夹的 fibo_1000.csv 文件,进行一些转为,保存为 sample.loom 文件,供后续流程。(这个时候的sample.loom 文件是后续一切分析的开始)
再次强调,使用conda安装的这个pyscenic,它依赖于一系列的python模块,就会在这里报错!比如我遇到的就是pandas的报错:
ImportError: cannot import name 'DtypeArg' from 'pandas._typing' (/home/x10/miniconda3/envs/pyscenic/lib/python3.7/site-packages/pandas/_typing.py)
不过,我使用了docker镜像的pyscenic,所以绕过了这个报错!
需要自己准备好如下所示的文件:
54M 7月 18 11:18 fibo_1000.csv
1.1G 7月 18 11:30 hg19-tss-centered-10kb-7species.mc9nr.feather
12K 7月 18 11:29 hs_hgnc_tfs.txt
99M 7月 18 11:29 motifs-v9-nr.hgnc-m0.001-o0.0.tbl
pyscenic 的3个步骤之 grn
代码如下所示,复制粘贴后运行即可:
pyscenic grn \
--num_workers 20 \
--output adj.sample.tsv \
--method grnboost2 \
sample.loom \
hs_hgnc_tfs.txt #转录因子文件,1839 个基因的名字列表
转录因子的基因列表文件下载 :pySCENIC/resources at master · aertslab/pySCENIC · GitHub
pyscenic 的3个步骤之 cistarget
代码如下所示,复制粘贴后运行即可:
同样的需要下载数据库文件,https://resources.aertslab.org/cistarget/
pyscenic ctx \
adj.sample.tsv \
hg38__refseq-r80__10kb_up_and_down_tss.mc9nr.feather \
--annotations_fname motifs-v9-nr.hgnc-m0.001-o0.0.tbl \
--expression_mtx_fname sample1.loom \
--mode "dask_multiprocessing" \
--output reg.csv \
--num_workers 3 \
--mask_dropouts
pyscenic 的3个步骤之 AUCell
这个步骤超级快,代码如下所示,复制粘贴后运行即可:
pyscenic aucell \
sample.loom \
reg.csv \
--output sample_SCENIC.loom \
--num_workers 3
sample_SCENIC.loom导入R里面进行可视化
首先再看看文章《Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma》,其降维聚类分群分群,并且拿出来fibo细胞亚群走这个转录因子分析,得到如下所示:
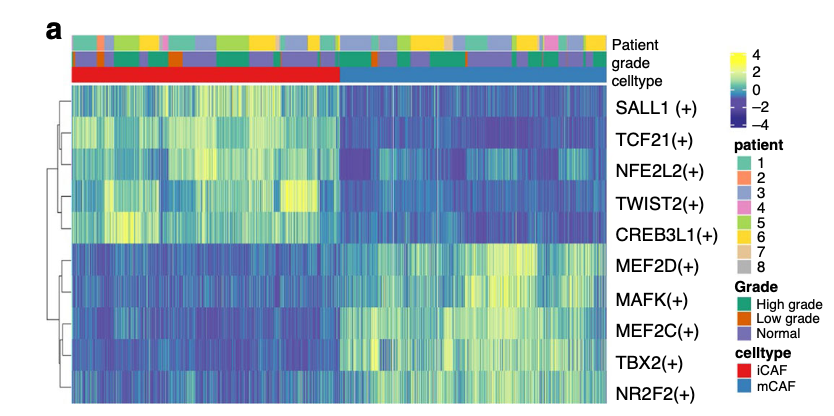
我们的复现分成2步,首先对进行降维聚类分群,代码如下所示:
rm(list = ls())
library(tidyverse)
library(Seurat)
library(data.table)
ct=fread('fibo_1000.csv',data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
ct=ct[,-1]
ct=t(ct)
fibo_sce <- CreateSeuratObject(counts = ct,
project = "fibo_1000" )
fibo_sce
#QC
fibo_sce[["percent.mt"]] <- PercentageFeatureSet(fibo_sce, pattern = "^MT-")
# Visualize QC metrics as a violin plot
VlnPlot(fibo_sce, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
fibo_sce <- subset(fibo_sce, subset = nFeature_RNA > 200 & nFeature_RNA < 5000 & percent.mt < 25)
#Normalization
fibo_sce <- NormalizeData(fibo_sce, normalization.method = "LogNormalize", scale.factor = 10000)
#高变基因选择
fibo_sce <- FindVariableFeatures(fibo_sce, selection.method = "vst", nfeatures = 2000)
#标准化
all.genes <- rownames(fibo_sce)
fibo_sce <- ScaleData(fibo_sce, features = all.genes)
#去除MT,重新进行标准化
fibo_sce <- ScaleData(fibo_sce, vars.to.regress = "percent.mt")
#PCA
fibo_sce <- RunPCA(fibo_sce, features = VariableFeatures(object = fibo_sce), verbose = FALSE)
#聚类
fibo_sce <- FindNeighbors(fibo_sce, dims = 1:20)
fibo_sce <- FindClusters(fibo_sce, resolution = 0.5)
#可视化
fibo_sce <- RunUMAP(fibo_sce, dims = 1:20)
fibo_sce<- RunTSNE(fibo_sce, dims = 1:20)
## after we run UMAP and TSNE, there are more entries in the reduction slot
str(fibo_sce@reductions)
DimPlot(fibo_sce, reduction = "umap", label = TRUE)
#查找marker基因
fibo_sce.markers <- FindAllMarkers(fibo_sce, only.pos = TRUE, min.pct = 0.25,
logfc.threshold = 0.25)
saveRDS(fibo_sce, "./fibo_1000.rds")
# 挑选合适的基因,进行可视化
出图如下:
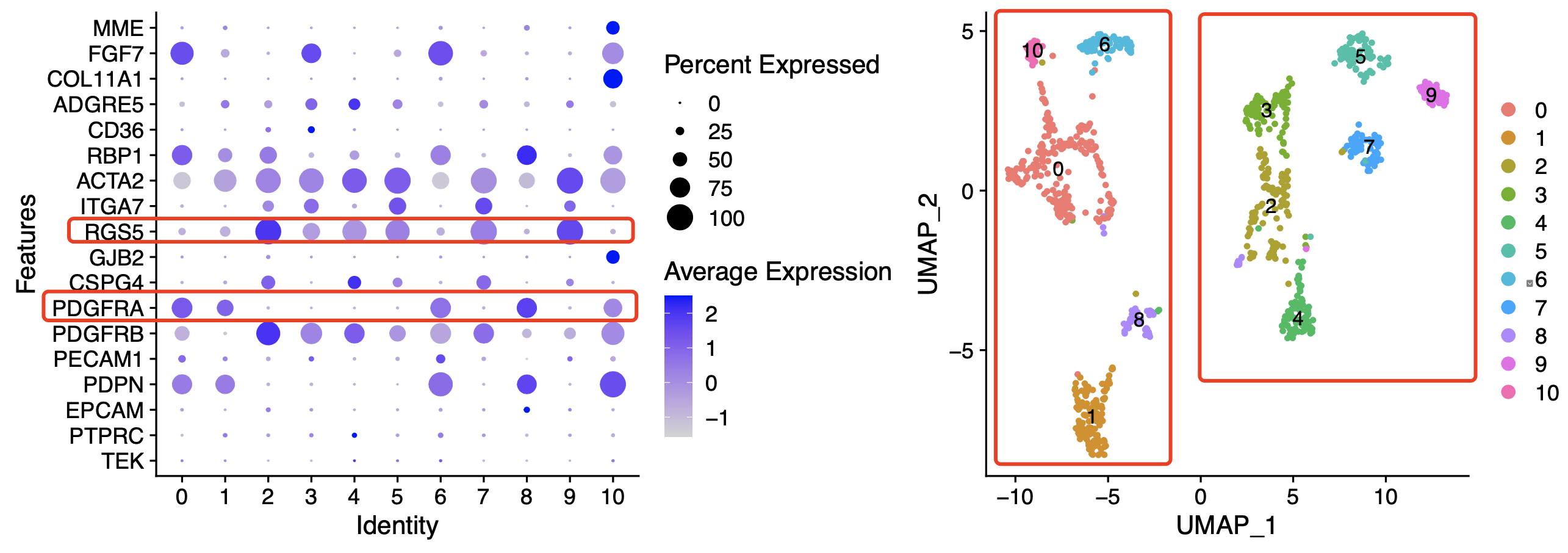
文章采取了RGS5和PDGFRA来区分iCAF和mCAF,我们姑且先按照作者的来!我们前面的降维聚类分群操作采取resolution = 0.5的时候确实得到了太多亚群,不过还是尊重作者,进行二分类!
rm(list = ls())
library(SCENIC)
packageVersion("SCENIC")
library(SCopeLoomR)
scenicLoomPath='sample_SCENIC.loom'
loom <- open_loom(scenicLoomPath)
# Read information from loom file:
regulons_incidMat <- get_regulons(loom, column.attr.name="Regulons")
regulons <- regulonsToGeneLists(regulons_incidMat)
regulonAUC <- get_regulons_AUC(loom, column.attr.name="RegulonsAUC")
sce=readRDS("./fibo_1000.rds")
sce
library(pheatmap)
n=t(scale(t( getAUC(regulonAUC[,] )))) # 'scale'可以对log-ratio数值进行归一化
n[n>2]=2
n[n< -2]= -2
n[1:4,1:4]
dim(n)
ac=data.frame(group= as.character( Idents(sce)))
n[1:4,1:4]
n=n[,colnames(n) %in% colnames(sce)]
rownames(ac)=colnames(n)
cg=read.table('choose_tf.txt')[,1]
cg
cg_n=n[rownames(n) %in% cg,]
pheatmap(cg_n,show_colnames =F,show_rownames = T,
annotation_col=ac)
table(ac$group)
# 尊重作者,进行二分类!
ac$group=ifelse(ac$group %in% c(2:5,7,9),'mCAF','iCAF')
pheatmap(cg_n,show_colnames =F,show_rownames = T,
annotation_col=ac)
pheatmap(cg_n,show_colnames =F,show_rownames = T,
annotation_col=ac,
filename = 'heatmap_choose_regulon.png')
dev.off()
出图如下:
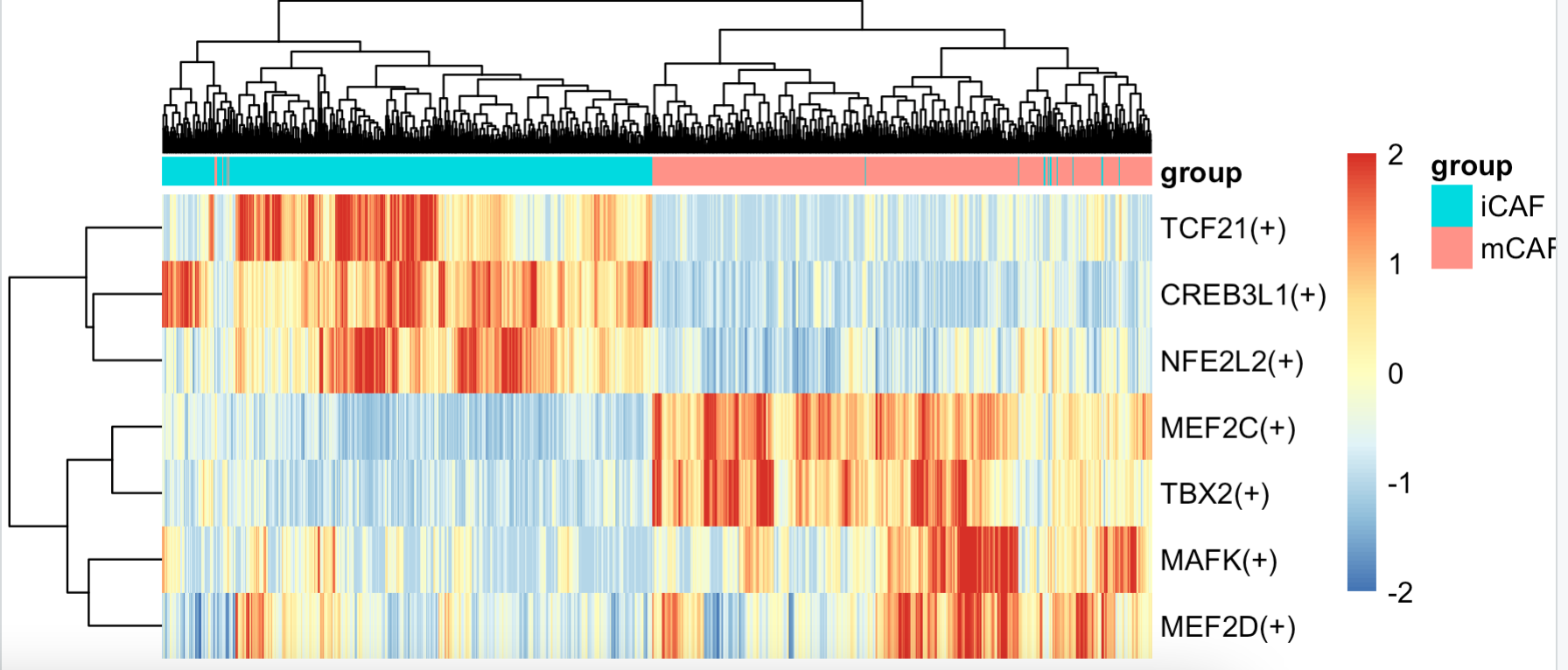
可以看到,作者的10个转录因子,在我们这个里面仅仅是剩下了 7 个,不知道是不是因为前面随机抽样1000个细胞导致的。虽然有转录因子的缺失,但是转录组因子的规律并没有变化,在iCAF和mCAF这个亚群特异性激活的转录因子保持原文的样子。