单细胞领域的一个高级分析是RNA速率分析,使用velocyto软件可以做,我们同样的把它区分为上下游分析。
上游分析需要在Linux操作环境里面,前面对10x的测序数据fq文件完成了 cellranger命令之后会有一个outputs文件夹。在该文件夹运行conda安装好的Python版本的velocyto软件即可,输出loom文件,供下游R里面操作。
安装自己的conda,每个用户独立操作
安装方法代码如下:
# 首先下载文件,20M/S的话需要几秒钟即可
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
# 接下来使用bash命令来运行我们下载的文件,记得是一路yes下去
bash Miniconda3-latest-Linux-x86_64.sh
# 安装成功后需要更新系统环境变量文件
source ~/.bashrc
安装好conda后需要设置镜像。
conda config --add channels r
conda config --add channels conda-forge
conda config --add channels bioconda
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/cloud/bioconda/
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/pkgs/main/
conda config --set show_channel_urls yes
使用conda安装velocyto的一些依赖
暂不支持conda直接安装 velocyto,将会在1.0版本使用 。
#需要一些依赖
conda create -n velocyto
conda activate velocyto
conda install samtools
conda install numpy scipy cython numba matplotlib scikit-learn h5py click
pip install pysam
# `PyPI`安装
pip install velocyto
# Successfully installed loompy-3.0.6 numpy-groupies-0.9.13 pandas-1.2.5 pytz-2021.1 velocyto-0.17.17
下载特定物种的特殊gtf文件
基因组注释文件主要是: GENCODE 或者Ensembl ; 不过, 我们这个单细胞转录组使用cellranger流程的话,需要重复数据的gtf文件,rmsk
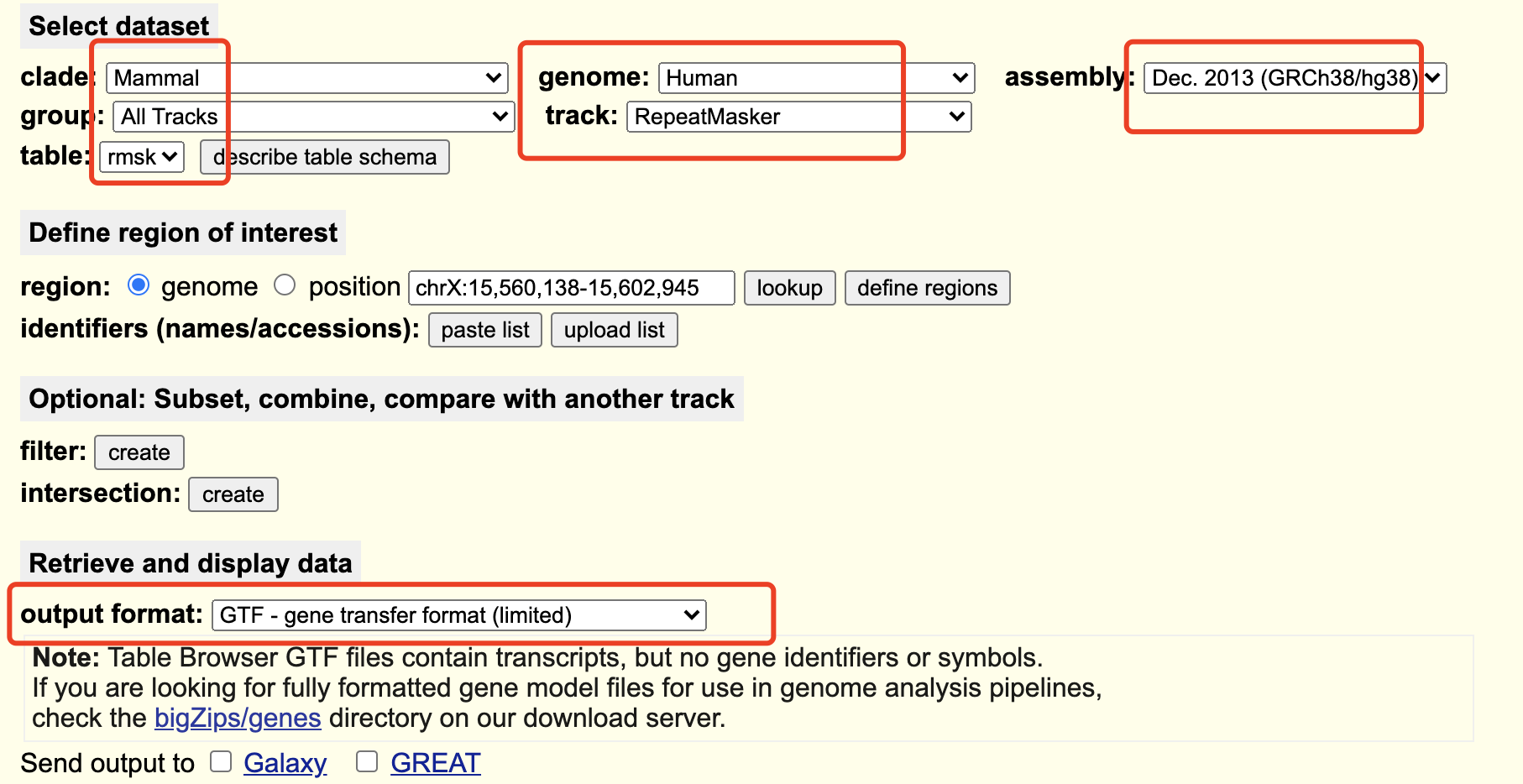
我们这里是 hg38_repeat_rmsk.gtf 文件,需要自己制作并且下载到指定工作目录!
从cellranger得到loom文件
这里需要 使用基于python的velocyto软件,它需要3个参数,其中两个是gtf文件,一个是前面cellranger命令的outputs目录哦,完整的命令如下:
rmsk_gtf=$HOME/pipeline/velocyto/hg38_repeat_rmsk.gtf # 从genome.ucsc.edu下载
cellranger_outDir=HSY-fushui # 前面cellranger命令的outputs目录
cellranger_gtf=$HOME/pipeline/refdata-gex-GRCh38-2020-A/genes/genes.gtf # 这个是cellranger官网提供的
ls -lh $rmsk_gtf $cellranger_outDir $cellranger_gtf
nohup velocyto run10x -m $rmsk_gtf $cellranger_outDir $cellranger_gtf &
# 如果是其它单细胞数据,可以换参数,比如run_smartseq2
理论上,这个一句话命令,就可以完成了python的velocyto软件并且拿到loom文件,但是,因为 samtools问题,上面的流程可能是会失败。
这个时候可以自行先运行 samtools sort 命令处理得到 cellsorted_possorted_genome_bam.bam 文件,如下所示的命令:
cd $cellranger_outDir/out
nohup samtools sort -@ 10 -t CB -O BAM -o cellsorted_possorted_genome_bam.bam possorted_genome_bam.bam &
# 这个 samtools sort 的 速度很快
# 下次就可以把前面的 velocyto run10x 重新跑一次,因为 samtools sort 命令已经是成功了。
# The file /home/---/outs/cellsorted_possorted_genome_bam.bam already exists.
# The sorting step w ill be skipped and the existing file will be used.
在同样的前面cellranger命令的outputs 文件夹目录下面,会输出一个 velocyto 文件夹,如下所示:
$ ls -lh velocyto/
158M 7月 3 22:38 HSY-fushui.loom
有了这个loom文件,后续就是回到R语言里面的统计可视化啦!
如果是多个10x样品都需要运行velocyto
首先,把全部的bam文件循环进行sort,代码如下:
ls */outs/possorted_genome_bam.bam|while read id;do new=${id/possorted_genome_bam.bam/cellsorted_possorted_genome_bam.bam}
echo $new
nohup samtools sort -@ 4 -t CB -O BAM -o $new $id &
done
这个代码难度有点高哦,需要精通Linux才能理解它。
同样的,然后可以批量走python的velocyto软件,代码如下所示:
rmsk_gtf=$HOME/pipeline/velocyto/hg38_repeat_rmsk.gtf # 从genome.ucsc.edu下载
#cellranger_outDir=HSY-fushui # 前面cellranger命令的outputs目录
cellranger_gtf=$HOME/pipeline/refdata-gex-GRCh38-2020-A/genes/genes.gtf # 这个是cellranger官网提供的
#ls -lh $rmsk_gtf $cellranger_outDir $cellranger_gtf
# 同样的一个简单的 循环即可
ls -d *-*|while read cellranger_outDir;do
nohup velocyto run10x -m $rmsk_gtf $cellranger_outDir $cellranger_gtf &
done
每个样品都会输出如下所示的loom文件:
158M 7月 19 17:39 HSY-fushui.loom
79M 7月 19 17:39 HSY-PBMC.loom
132M 7月 19 17:39 HSY-yi.loom
163M 7月 19 17:39 HSY-yuan.loom
246M 7月 19 17:39 LS-Endo-Pro.loom
139M 7月 19 17:39 LS-PBMC-Pro.loom
211M 7月 19 17:39 RDX-PBMC.loom
205M 7月 19 17:39 RDX-yuan.loom
123M 7月 19 17:39 WY-PBMC.loom
91M 7月 19 17:39 WY-yi.loom
136M 7月 19 17:39 WY-yuan.loom
253M 7月 19 17:39 YF-fushui.loom
188M 7月 19 17:39 YF-PBMC.loom
135M 7月 19 17:39 YF-yi.loom
148M 7月 19 17:39 YF-yuan.loom
243M 7月 19 17:39 YX-Endo-Decidu.loom
128M 7月 19 17:39 YX-PBMC-Decidu.loom
95M 7月 19 17:39 ZZX-PBMC.loom
123M 7月 19 17:39 ZZX-yuan-2.loom
拿到了这么多 loom文件,就可以进行下游 velocyto.R 这个R包进行后续统计可视化啦!明天我们就讲解velocyto.R 这个R包用法!
关于RNA velocity (gene expression trajectory)
RNA velocity是基于真实的转录动力学,可用于细胞基因表达的动态分化的研究。
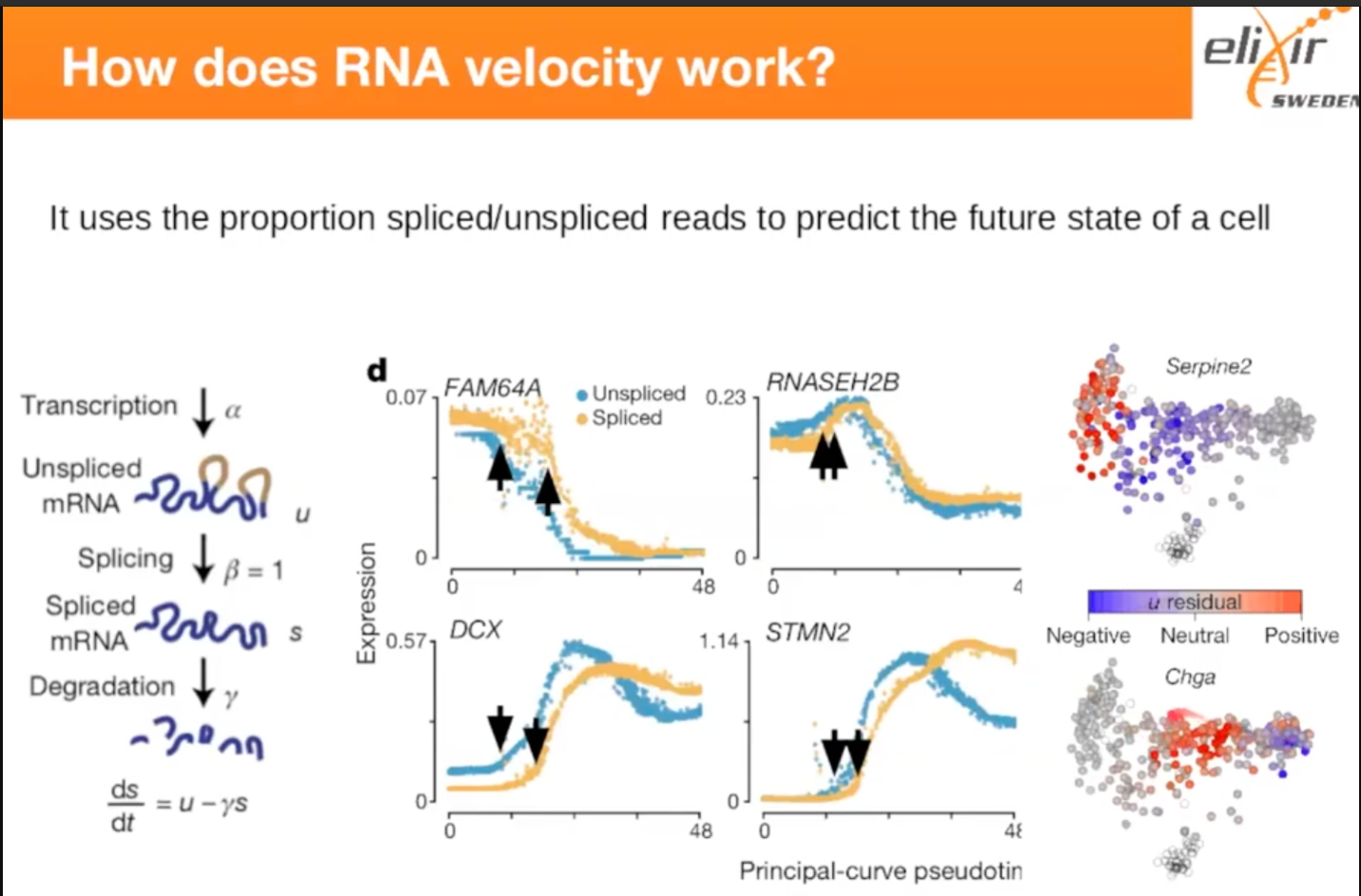
如上左图,刚转录出的mRNA包含外显子和内含子,经过splicing切除内含子后,得到用于编码 蛋白的spliced mRNA。spliced mRNA的丰度由未成熟mRNA的splicing速度和降解速率共同决 定。如上中图:每个点代表一个细胞,在拟时间轴上,未经过剪切的mRNA的出现始终早于经 过剪切的mRNA。如上右图:红色代表未经过剪切的mRNA,蓝色代表经过剪切的mRNA,可以 看出,这些细胞的应该是从左往右分化的,因此Velocity可以用于定义轨迹的起点分支和终点。 也就是说,Velocity可以在不知发育过程的前提下,预测谱系的方向(如下图)。
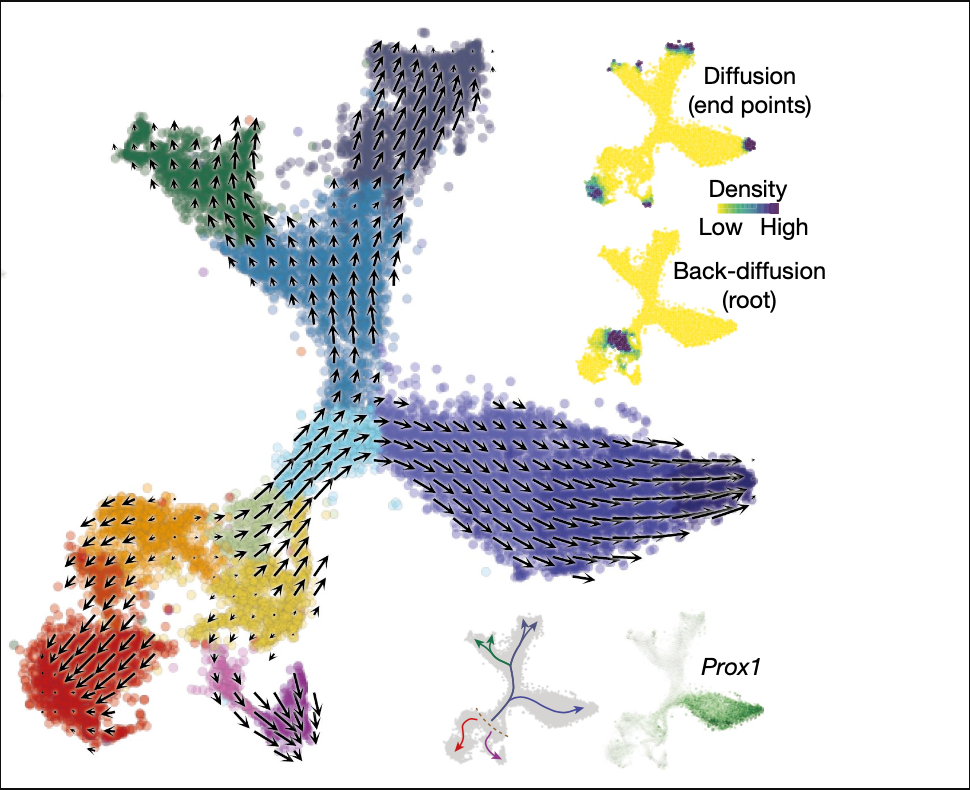
Velocity可以用于周期的轨迹