昨天我们给《单细胞天地》的交流群的粉丝提问,关于 FindMarkers与AverageExpression 两个函数的差异,做出来一个简单的示意图解释,见:[这算是不一样吗](https://mp.weixin.qq.com/s/s25DLc-tj0lPAcsPurn89Q),拿到了两个函数,FindMarkers与AverageExpression的各自计算的差异倍数的散点图, 可以看到两次计算结果几乎是没有差异,这样的0.91的相关性已经是非常好了。
但是仍然是会有不少人,不依不饶,一定要得到一模一样的结果,我就在《单细胞天地》的交流群号召大家参与创作,其中山东大学的王晶给出来了自己的解释,非常棒!
我们进一步深究,选取PPBP基因作为研究对象,如下所示:
av['PPBP',]
## DC Platelet diff
## PPBP 0.0674509 442.2696 7.879211
发现其在血小板内的表达量很高,差异变化的倍数取了log2,都有 7.879211怎么高!
真实情况呢?
hist(as.matrix(sce@assays$RNA@data)['PPBP',Idents(sce)=='Platelet'])
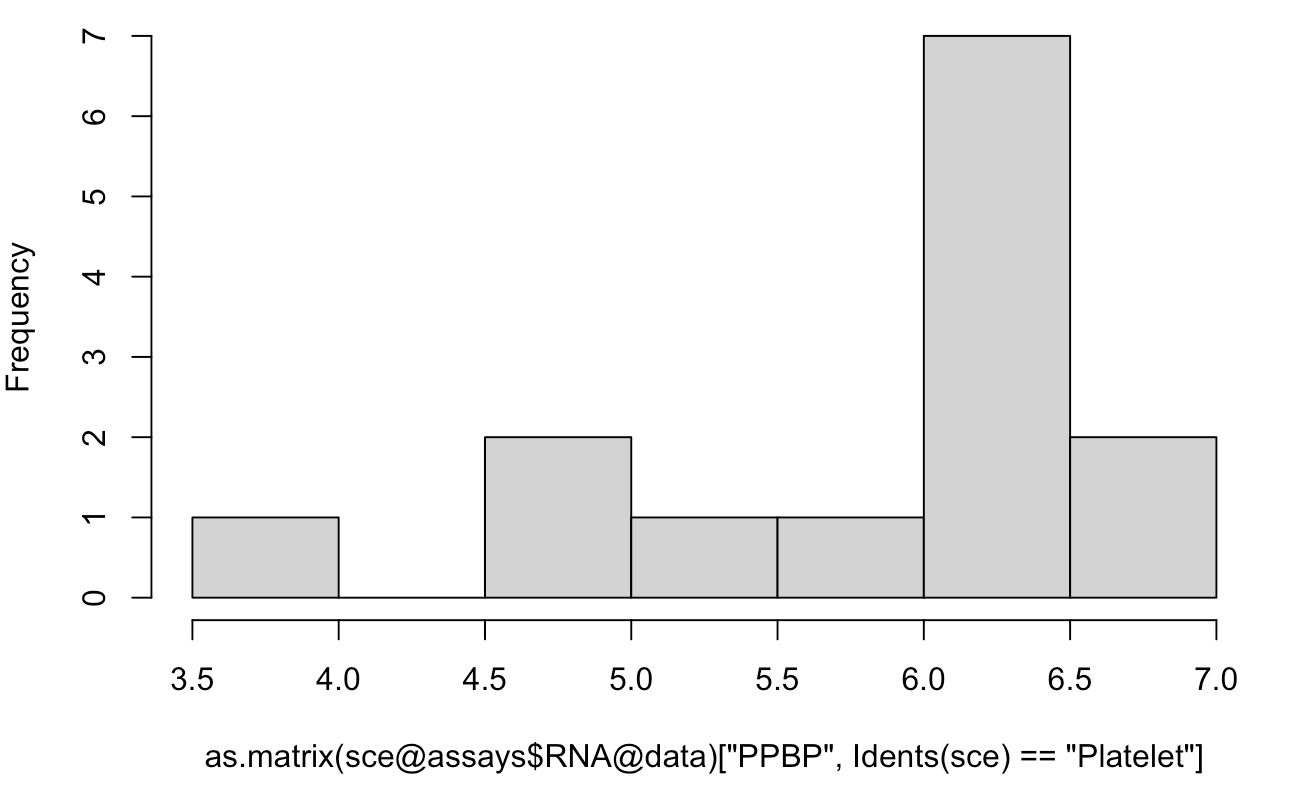
可以看到大部分的表达量都及集中在6.0到6.5,这显然是log转换后的数据,与AverageExpression给出的平均表达量442.2696对应不上,因为我们看的是 sce@assays$RNA@data 里面的矩阵。
查询AverageExpression函数的帮助文档,注意到一句话:
If slot is set to ‘data’, this function assumes that the data has been log normalized and therefore feature values are exponentiated prior to averaging so that averaging is done in non-log space.
意思是说如果slot按照默认设定为’data’,在进行平均值运算之前需要先进行去log化。我们可以比较一下多个矩阵:
exp1 <- as.matrix(sce@assays$RNA@data)
exp2 <- as.matrix(sce@assays$RNA@counts)
exp3 <- as.matrix(sce@assays$RNA@scale.data)
对这3个矩阵进行探索:
group <- Idents(sce)
test_exp2 <- data.frame(exp1 = exp1['PPBP',],
exp2 = exp2['PPBP',],
exp3 = exp3['PPBP',],
group = group)
test_exp2$exp1_nonlog <- exp(test_exp2$exp1)-1
test_exp2 <- test_exp2[,c(1,5,2,3,4)]
head(test_exp2)
## exp1 exp1_nonlog exp2 exp3 group
## AAGATTACCGCCTT-1 0.000000 0.0000 0 -0.1356634 DC
## AAGCCATGAACTGC-1 0.000000 0.0000 0 -0.1295316 DC
## AATTACGAATTCCT-1 0.000000 0.0000 0 -0.1377150 DC
## ACCCACTGGTTCAG-1 5.983139 395.6835 22 10.0000000 Platelet
## ACCCGTTGCTTCTA-1 0.000000 0.0000 0 -0.1300184 DC
## ACCTGAGATATCGG-1 4.933652 137.8858 21 9.4924156 Platelet
colMeans(test_exp2[group == 'Platelet',1:4])
## exp1 exp1_nonlog exp2 exp3
## 5.851007 442.269608 40.785714 9.696587
colMeans(test_exp2[group == 'DC',1:4])
## exp1 exp1_nonlog exp2 exp3
## 0.03593983 0.06745090 0.03125000 -0.06931771
重点看exp1_nonlog一列,可以看到对’data’ slot内的数据去log化后,DC组和Platelet组的平均表达量与AverageExpression函数计算的结果一致了。
如果挖掘到这里就结束了,那就没什么意思了,我们还要继续探究FindMarkers的具体计算策略:
“wilcox” : Identifies differentially expressed genes between two groups of cells using a Wilcoxon Rank Sum test (default)
查询帮助文档得知默认是用Wilcox检验。
那么我们直接手动计算一下PPBP的P value和log2FC。
注意,计算log2FC必须是用non-log space下的数据。
wilcox.test(exp1~group,test_exp2)
##
## Wilcoxon rank sum test with continuity correction
##
## data: exp1 by group
## W = 0, p-value = 1.511e-10
## alternative hypothesis: true location shift is not equal to 0
mean1 <- mean(test_exp2$exp1_nonlog[group == 'DC'])
mean2 <- mean(test_exp2$exp1_nonlog[group == 'Platelet'])
log2(mean2+1)-log2(mean1+1)
## [1] 8.697871
diff['PPBP',]
## p_val avg_log2FC pct.1 pct.2 p_val_adj
## PPBP 1.511444e-10 8.697871 1 0.031 2.072795e-06
手动计算的p值为1.511e-10,log2FC为8.697871,与FindMarkers自动计算的结果完全一致。
那么正确的利用AverageExpression函数计算avg_log2FC的方法为:
av$diff2 <- log2(av[,2]+1) - log2(av[,1]+1)
所以重点是 log2的时候,先加上一个1作为虚拟表达量。
再次 验证一下:
comp=data.frame(FindMarkers=diff[ids,'avg_log2FC'],
AverageExpression=av[ids,'diff'],
AE_update=av[ids,'diff2'])
head(comp)
## FindMarkers AverageExpression AE_update
## 1 8.697871 7.879211 8.697871
## 2 7.048021 6.244134 7.048021
## 3 5.126740 5.830073 5.126740
## 4 5.127601 5.830686 5.127601
## 5 5.858386 6.347674 5.858386
## 6 6.609978 6.875016 6.609978
ggscatter(comp, x = "FindMarkers", y = "AE_update",
color = "black", shape = 21, size = 3, # Points color, shape and size
add = "reg.line", # Add regressin line
add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
conf.int = TRUE, # Add confidence interval
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.sep = "\n")
)
可以看到,相关性接近于 1
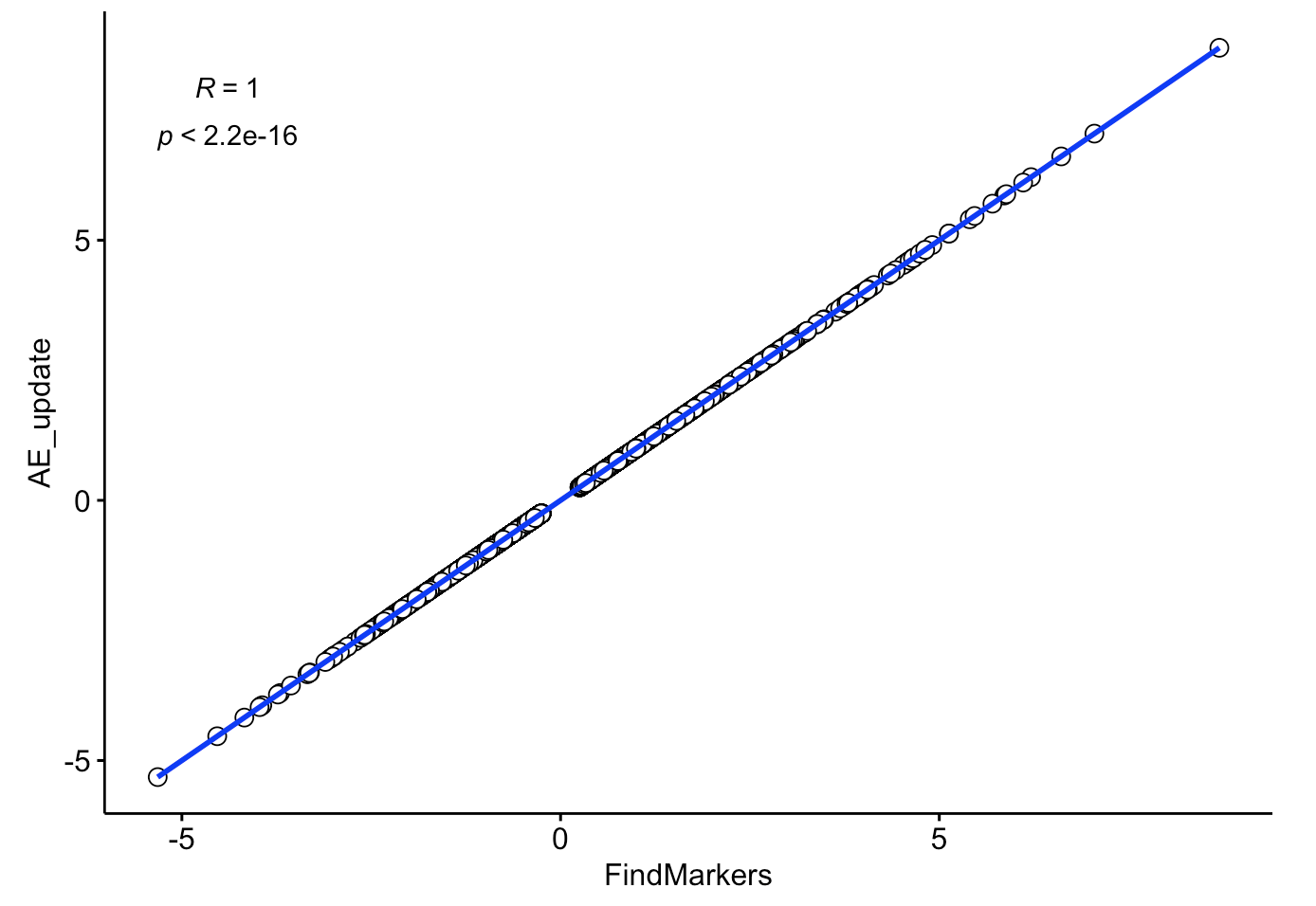
是不是非常棒啊!