前面我们针对TCGA数据库全部的癌症的表达量矩阵批量运行 estimate,而且得到了estimate的两个打分值,可以发现可以看到不同癌症内部的estimate的两个打分值是有异质性的。
其实这个estimate分析,它本质上是ssGSEA,这个分析依赖于同一个样品内部基因的排序,所以tpm或者fpkm的归一化会改变样品内基因的排序,所以结果会有差异!
现在我们就来具体探索一下这些差异!
首先看看独立癌症estimate结果和全癌症样本队列运行结果
如果要全癌症样本队列运行estimate的结果,只需针对前面的seurat对象运行 estimate 即可,代码如下所示:
###### step4: 针对前面的seurat对象运行 estimate ######
library(Seurat)
load(file = 'sce.Rdata')
gp=substring(colnames(sce),14,15)
table(gp)
sce@meta.data$gp=gp
pd_mat=sce@assays$RNA@counts
dat=log2(edgeR::cpm(pd_mat)+1)
pro='estimate_for_seurat'
scores=estimate_RNAseq(dat,pro)
head(scores)
scores$group= sce@meta.data$gp
scores$type= sce@meta.data$group
save(scores,file = file.path('estimate_results',
paste0('estimate_RNAseq-score-for-',
pro,'.Rdata')))
比较不同 estimate 结果
# 然后批量载入各自 estimate 结果
fs=list.files('estimate_results/',
pattern = 'estimate_RNAseq-score-forTCGA')
fs
lapply(fs, function(x){
# x=fs[1]
pro=gsub('.Rdata','',
gsub('estimate_RNAseq-score-forTCGA-','',x))
print(pro)
load(file = file.path('estimate_results/',x))
df=cbind(scores[,1:3],
all_scores[rownames(scores) ,1:3])
kp=apply(df, 2,sd) > 0
df=df[,kp]
m=cor(df)
#pheatmap::pheatmap( cor(df))
pheatmap::pheatmap( cor(df),
filename = file.path('estimate_results',
paste0('cor-by-merge-and-single-',pro,'.pdf')))
})
从上面代码看,我们是把33个癌症内部,都做了两次运行 estimate 结果的对比,随便拿出来一个癌症看相关性:
> options(digits=2)
> m
StromalScore ImmuneScore ESTIMATEScore StromalScore.1 ImmuneScore.1 ESTIMATEScore.1
StromalScore 1.00 0.82 0.94 1.00 0.82 0.94
ImmuneScore 0.82 1.00 0.96 0.82 1.00 0.96
ESTIMATEScore 0.94 0.96 1.00 0.94 0.96 1.00
StromalScore.1 1.00 0.82 0.94 1.00 0.82 0.94
ImmuneScore.1 0.82 1.00 0.96 0.82 1.00 0.96
ESTIMATEScore.1 0.94 0.96 1.00 0.94 0.96 1.00
需要热图可视化:
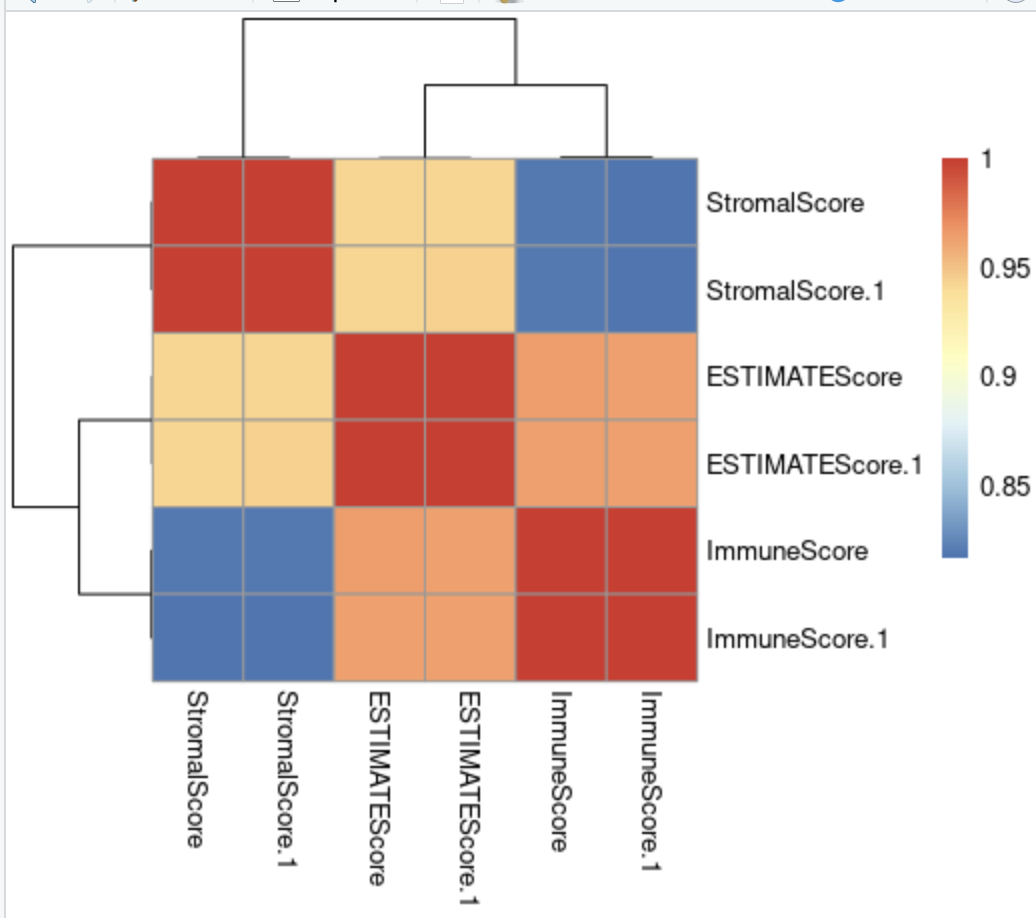
可以看到合并TCGA的33个癌症一起走estimate,与在每个癌症内部独立走estimate,得到的结果是没有差异的。所以estimate算法本身的样品独立性的,它关心的仅仅是每个样品内部的基因排序。其实就是ssGSEA算法啦!
那么使用ssGSEA代替estimate呢
代码如下所示:
rm(list=ls())
options(stringsAsFactors = F)
library(stringr)
library(data.table)
library(Seurat)
load(file = 'sce.Rdata')
gp=substring(colnames(sce),14,15)
table(gp)
sce@meta.data$gp=gp
pd_mat=sce@assays$RNA@counts
dat=log2(edgeR::cpm(pd_mat)+1)
pro='ssGSEA_for_seurat'
gmtFile=system.file("extdata", "SI_geneset.gmt",
package="estimate")
gmtFile
library(GSVA)
library(limma)
library(GSEABase)
library(data.table)
geneSet=getGmt(gmtFile,
geneIdType=SymbolIdentifier())
geneSet
StromalGenes=geneSet[[1]]@geneIds
ImmuneGenes=geneSet[[2]]@geneIds
# 参考:http://www.bio-info-trainee.com/6602.html
geneSet
as.matrix(dat)[1:4,1:4]
ssgseaScore=gsva(as.matrix(dat), geneSet,
method='ssgsea', kcdf='Gaussian',
abs.ranking=TRUE)
ssgseaScore=as.data.frame(t(ssgseaScore))
head(ssgseaScore)
save(ssgseaScore,file = file.path('estimate_results',
paste0('ssgseaScore-for-',
pro,'.Rdata')))
接下来就可以比较ssGSEA的打分值和estimate :
> head(scores)
StromalScore ImmuneScore ESTIMATEScore group type
TCGA.OR.A5JP.01A -998 -1415 -2413 01 ACC
TCGA.OR.A5JG.01A -1116 -963 -2079 01 ACC
TCGA.OR.A5K1.01A -905 -672 -1576 01 ACC
TCGA.OR.A5JR.01A -1192 -670 -1862 01 ACC
TCGA.OR.A5KU.01A -1171 -1483 -2655 01 ACC
TCGA.OR.A5L9.01A -461 79 -381 01 ACC
> rownames(ssgseaScore)=gsub('-','.', rownames(ssgseaScore))
> head(ssgseaScore)
StromalSignature ImmuneSignature
TCGA.OR.A5JP.01A -0.066 -0.139
TCGA.OR.A5JG.01A -0.091 -0.074
TCGA.OR.A5K1.01A -0.060 -0.023
TCGA.OR.A5JR.01A -0.099 -0.015
TCGA.OR.A5KU.01A -0.111 -0.168
TCGA.OR.A5L9.01A 0.011 0.100
可以看到,如果肉眼看,会觉得两个结果似乎是完全不一样的,因为根本就不是一个数量级!
但是,好玩的地方在于,这两个完全不一样的打分结果矩阵,scores和ssgseaScore的相关性是1 :
> identical(rownames(scores),
+ rownames(ssgseaScore) )
[1] TRUE
> cor(scores$StromalScore,ssgseaScore$StromalSignature)
[1] 1
> cor(scores$ImmuneScore,ssgseaScore$ImmuneSignature)
[1] 1
当然了,也可以可视化,代码如下:
df=cbind(ssgseaScore,scores)
head(df)
library(ggpubr)
library(papatchwork)
p1=ggscatter(df, x = "StromalSignature", y = "StromalScore",
color = "black", shape = 21, size = 3, # Points color, shape and size
add = "reg.line", # Add regressin line
add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
conf.int = TRUE, # Add confidence interval
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.sep = "\n")
)
p1
p2=ggscatter(df, x = "ImmuneSignature", y = "ImmuneScore",
color = "black", shape = 21, size = 3, # Points color, shape and size
add = "reg.line", # Add regressin line
add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
conf.int = TRUE, # Add confidence interval
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.sep = "\n")
)
p2
p1+p2
出图如下:
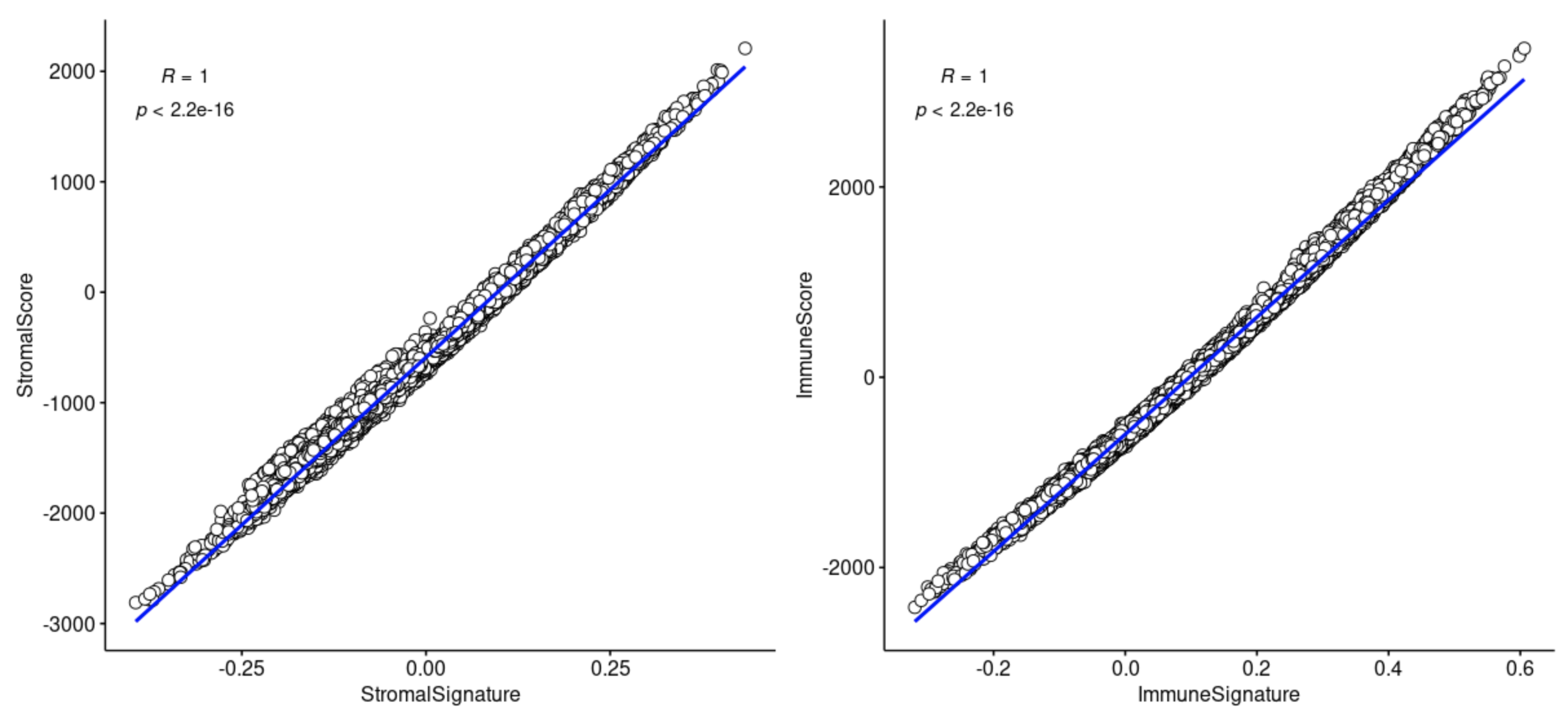
同样的说明了两个算法,得到结果是没有本质上的差异,仅仅是数值进行统计变化,并不影响它们的相关性!