我们在前面使用3个教程来说明肿瘤微环境,,是一个目前比较流行的方法 estimate看基质和免疫细胞比例 ,来源于2013数据挖掘文章,作者就整理了两个基因集来根据表达量矩阵使用estimate方法去量化肿瘤样品里面的基质细胞和免疫细胞的比例。目录是:
- estimate的两个打分值本质上就是两个基因集的ssGSEA分析
- 针对TCGA数据库全部的癌症的表达量矩阵批量运行estimate
- 不同癌症内部按照estimate的两个打分值高低分组看蛋白编码基因表达量差异
实际上, 这个方法还是过于粗糙了,肿瘤微环境的复杂程度,远不止基质和免疫细胞简单的归类。我随手查了一个比较新的综述文章:《Tumor microenvironment complexity and therapeutic implications at a glance》,链接是https://biosignaling.biomedcentral.com/articles/10.1186/s12964-020-0530-4,感兴趣的可以自己研读:
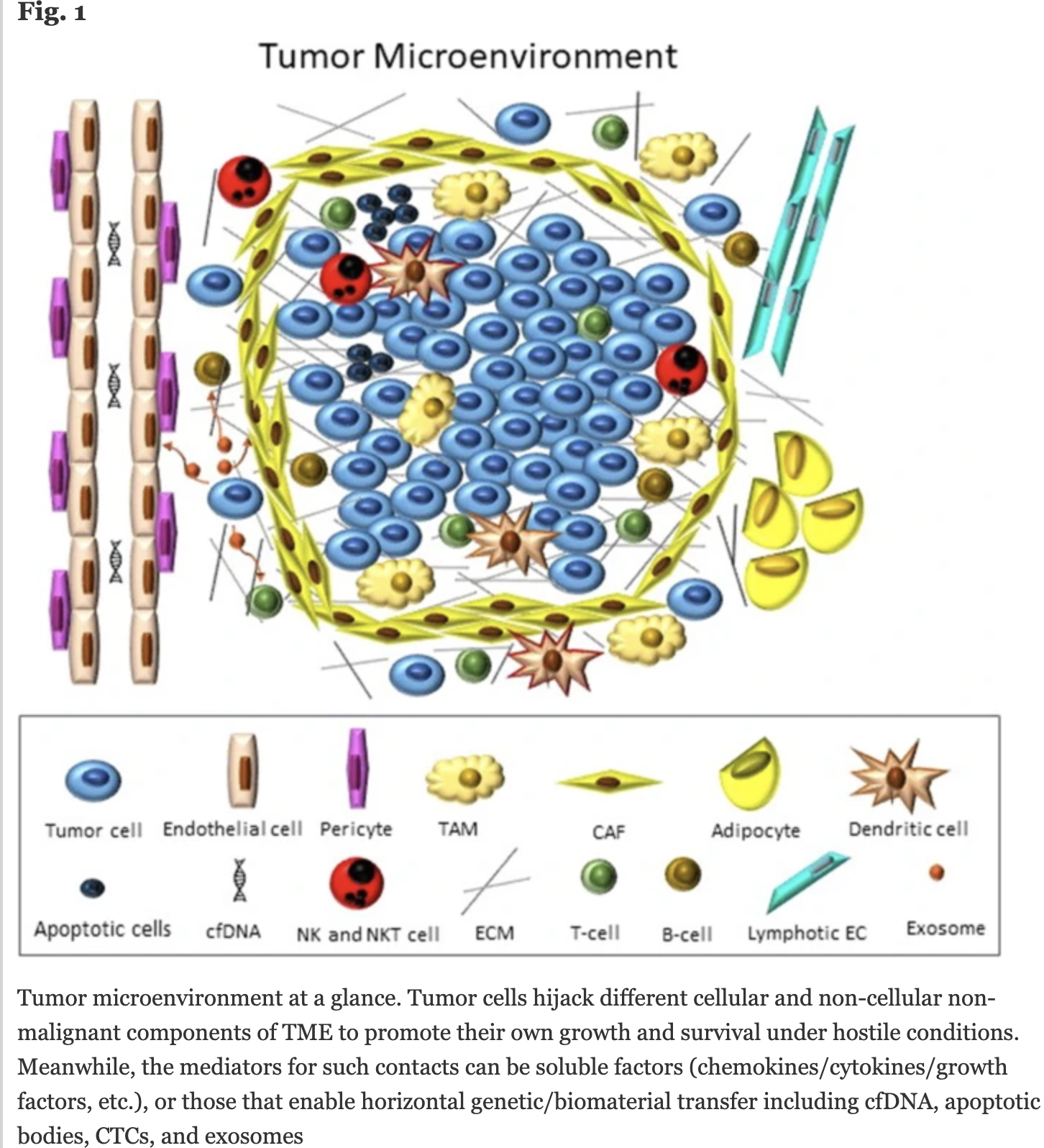
可以看到,免疫细胞有淋巴系的T,nk和b细胞,还有髓系的dc和TAM等等,统一的免疫细胞比例肯定是很难说明肿瘤真实的免疫情况,因为不同免疫细胞的作用并不一样。
但是咱们的TCGA数据库的转录组测序数据毕竟是bulk转录组测序,病人的肿瘤样品里面虽然是混合了各种各样的免疫细胞,但是在数据层面被混杂成为了一个样品,并不是单细胞测序,所以并没有细胞比例信息。这个时候仍然是需要借助于算法啦,有一个2019的综述文章:《Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology》比较了常见的免疫细胞比例推断工具的表现:

但是你,工具的表现并不能决定它的流行程度,流行与否主要是靠传播,这个时候CIBERSORT就一马当先了,CIBERSORT是2015年在Nature Methods发表的一个方法,工具在: (http://cibersort.stanford.edu)., 早在2016就有文章使用它进行数据挖掘了,比如. Patterns of Immune Infiltration in Breast Cancer and Their Clinical Implications: A Gene-Expression-Based Retrospective Study. PLOS Medicine, 作者研究团队利用CIBERSORT算法推断解析了11,000个乳腺癌(组织转录组芯片或是RNAseq,包括GEO和TCGA)中的22种免疫细胞的占比。
使用CIBERSORT算法推断全部tcga样品的免疫细胞比例
这里我们直接对seurat对象走 CIBERSORT,如果你还不知道下面的代码里面的seurat对象是如何产生的,见:前面的教程:大样本量多分组表达量矩阵分析你难道没想到单细胞吗:
rm(list=ls())
options(stringsAsFactors = F)
library(stringr)
library(data.table)
library(preprocessCore)
library(parallel)
library(e1071)
library(dplyr)
library(tidyr)
library(tidyverse)
library(Seurat)
load(file = 'sce.Rdata')
gp=substring(colnames(sce),14,15)
table(gp)
sce@meta.data$gp=gp
pd_mat=sce@assays$RNA@counts
dat=log2(edgeR::cpm(pd_mat)+1)
pro='CIBERSORT_for_seurat'
# 只能说假装是 tpm,去搞基因长度信息很麻烦
exp_tpm=dat
exp_tpm[1:4,1:4]
source("CIBERSORT.R")
load('lm22.rda')
ciber=CIBERSORT(dat)
cibersort_raw <- read.table("CIBERSORT-Results.txt",header = T,sep = '\t') %>%
rename("Patients" = "Mixture") %>%
select(-c("P.value","Correlation","RMSE"))
dim(cibersort_raw)
sce
cibersort_raw$group= sce@meta.data$gp
cibersort_raw$type= sce@meta.data$group
pro='seurat'
save(cibersort_raw,
file = file.path('CIBERSORT_results',
paste0('cibersort_raw-for-',pro,'.Rdata')))
核心的代码就3句话:
source("CIBERSORT.R")
load('lm22.rda')
ciber=CIBERSORT(dat)
需要一个脚本, 就是 CIBERSORT.R,在其官网或者我们的教程配套网盘里面都有,见解析CIBERSORT使用SVM算法实现去卷积,
其次是lm22.rda,这个是我教程配套网盘里面的,见解析CIBERSORT使用SVM算法实现去卷积,
最后只需要给一个表达量矩阵即可,运行 CIBERSORT(dat) 函数,就可以拿到全部的结果啦!
可视化CIBERSORT算法推断的免疫细胞比例
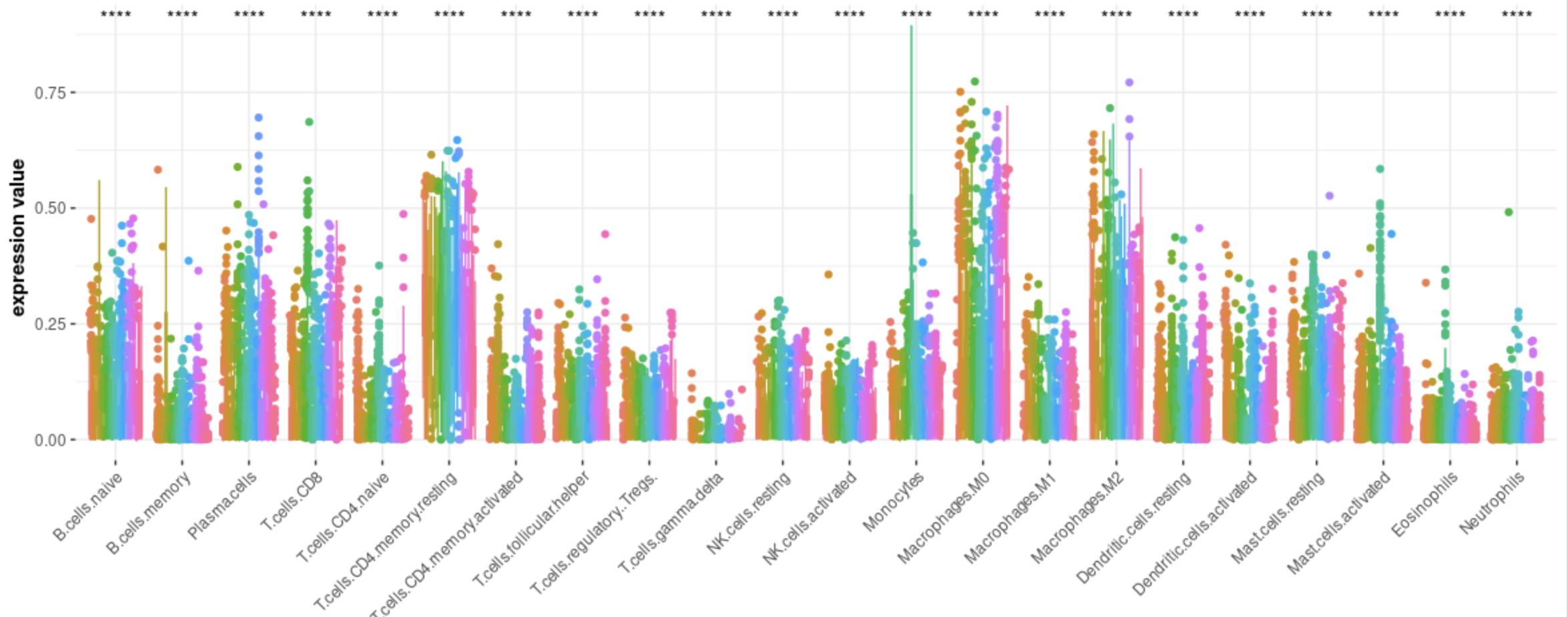
首先可以看到不同癌症免疫细胞比例差异很大:
其实,我们可以在: https://gdc.cancer.gov/about-data/publications/panimmune 找到 TCGA.Kallisto.fullIDs.cibersort.relative.tsv文件, 它的下载地址是 https://api.gdc.cancer.gov/data/b3df502e-3594-46ef-9f94-d041a20a0b9a
我们可以比较一下自己计算的免疫细胞比例和这个TCGA官方的差异:
cibersort_all <- read.table("CIBERSORT-Results.txt",header = T,sep = '\t') %>%
rename("Patients" = "Mixture") %>%
select(-c("P.value","Correlation","RMSE"))
cibersort_all[1:4,1:4]
library(data.table)
TCGA.Kallisto.fullIDs.cibersort = fread('TCGA.Kallisto.fullIDs.cibersort.relative.tsv',data.table = F)
TCGA.Kallisto.fullIDs.cibersort[1:4,1:4]
我们两个结果的样品id不一致:
> TCGA.Kallisto.fullIDs.cibersort[1:4,1:4]
SampleID CancerType B.cells.naive B.cells.memory
1 TCGA.OR.A5JG.01A.11R.A29S.07 ACC 0.000000e+00 0.04852871
2 TCGA.OR.A5LG.01A.11R.A29S.07 ACC 7.168876e-03 0.01112538
3 TCGA.OR.A5JD.01A.11R.A29S.07 ACC 2.342245e-05 0.01460659
4 TCGA.OR.A5LH.01A.11R.A29S.07 ACC 4.729908e-02 0.03817952
> cibersort_all[1:4,1:4]
Patients B.cells.naive B.cells.memory Plasma.cells
1 TCGA-OR-A5JP-01A 0.07674056 0 0
2 TCGA-OR-A5JG-01A 0.00000000 0 0
3 TCGA-OR-A5K1-01A 0.03410138 0 0
4 TCGA-OR-A5JR-01A 0.03491271 0 0
所以需要进行简单的转换,然后计算相关性:
identical(colnames(cibersort_all)[2:22],
colnames(TCGA.Kallisto.fullIDs.cibersort)[3:23])
pid=gsub('[.]','-',substring(TCGA.Kallisto.fullIDs.cibersort$SampleID,1,16))
head(pid)
table(cibersort_all$Patients %in% pid)
pos=match(cibersort_all$Patients,pid)
TCGA.Kallisto.fullIDs.cibersort=TCGA.Kallisto.fullIDs.cibersort[pos,]
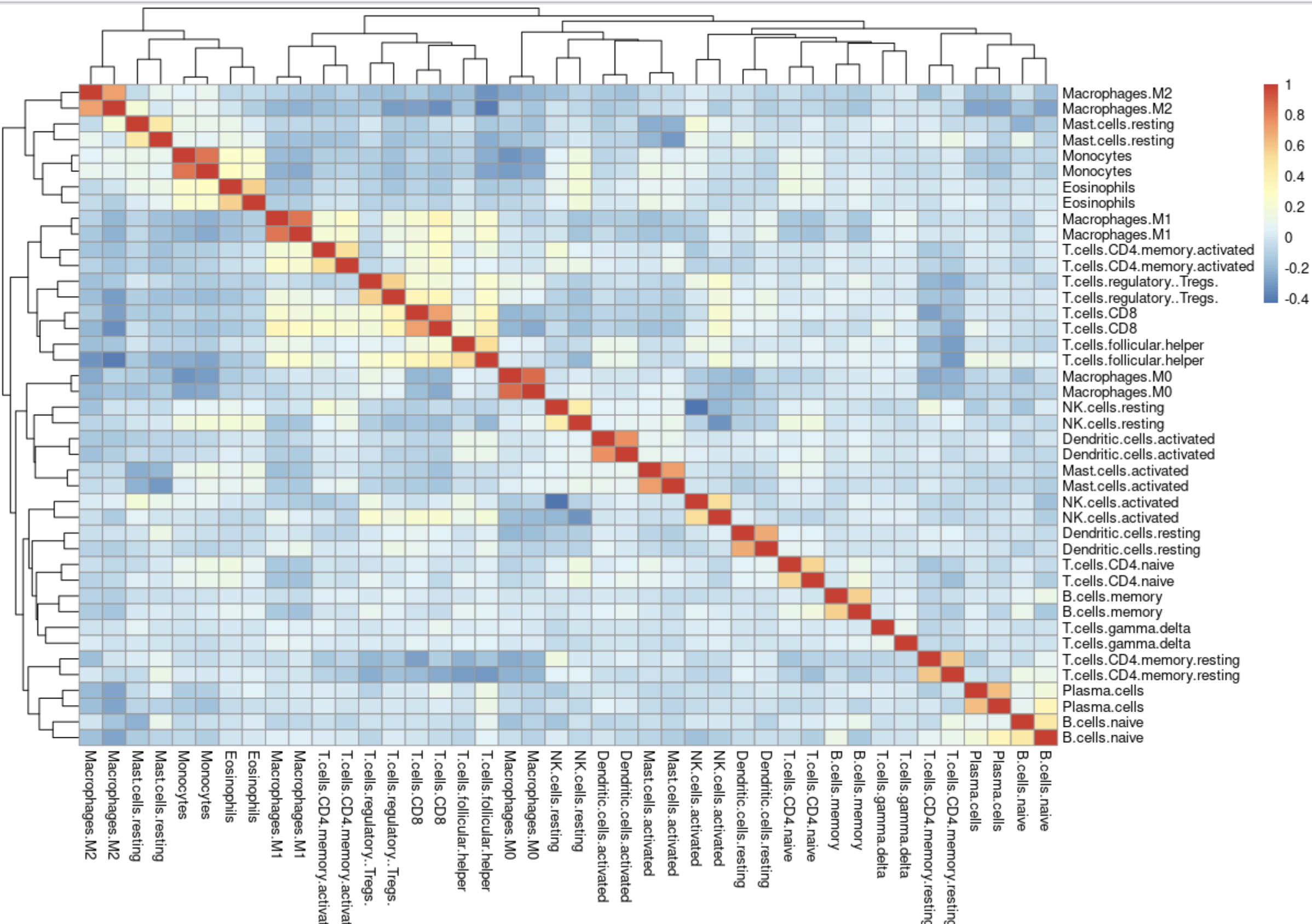
pheatmap::pheatmap(cor(
cbind(cibersort_all[2:22],
TCGA.Kallisto.fullIDs.cibersort[,3:23])
))
可以看到,虽然我跟TCGA官方的表达量矩阵形式不一样,但是我们计算得到的免疫细胞比例基本上一致。
前面我们提出来了疑问,就是针对RNA-seq数据,我们创造性的使用logCPM这样的表达量矩阵,进行estimate或者CIBERSORT算法流程(必然与tpm或者fpkm矩阵结果不一致哦!)
如果是根据具体某个基因表达量把病人分组,那么tpm或者fpkm跟我们的logCPM值并不会有区别,就是说差异分析,生存分析都没有问题。
但是呢,如果我们进行estimate或者CIBERSORT算法,都依赖于同一个样品内部基因的排序,所以tpm或者fpkm的归一化会改变样品内基因的排序,所以结果会有差异!
但是这个差异又是在可接受范围内!