我们在前面的教程:不是maf格式的somatic突变数据就没办法读入到maftools了么,提到了假如小伙伴们是在 https://xenabrowser.net/datapages/ ,找到 GDC TCGA Breast Cancer (BRCA) (20 datasets) ,数据库提供了4种somatic突变的maf文件供下载,somatic mutation (SNPs and small INDELs) ,一般来说我们选择GATK团队出品的MuTect2 软件拿到的somatic突变数据文件;
- MuTect2 Variant Aggregation and Masking (n=986) GDC Hub
这个时候呢,你会发现下载的突变数据是tsv格式,并不是maf格式,读入这样的tsv格式的肿瘤突变是信息需要一定技巧哦!可以再次学习前面的教程:不是maf格式的somatic突变数据就没办法读入到maftools了么。
其实呢,TCGA 的各个癌症的突变信息太分散而且版本一直在迭代,大家的下载源头千奇百怪。几年前我就系统性的介绍过:TCGA的pan-caner资料大全(以后挖掘TCGA数据库就用它) 还专门指出了癌症的somatic突变的maf文件问题:TCGA数据库maf突变资料官方大全。因为TCGA计划跨时太长,这些年找somatic变异的软件也很多,所以TCGA团队下功夫在计划结束后(April 2018)完整的系统性的整理了最后的somatic突变数据。依托于文章:Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines March 201810.1016/j.cels.2018.03.002
统一下载这个文件即可:https://api.gdc.cancer.gov/data/1c8cfe5f-e52d-41ba-94da-f15ea1337efc
rm(list = ls())
require(maftools)
laml=read.maf('mc3.v0.2.8.PUBLIC.maf.gz')
laml
save(laml,file = 'mc3.Rdata')
rm(list = ls())
require(maftools)
load(file = 'mc3.Rdata')
plotmafSummary(maf = laml,
rmOutlier = TRUE, addStat = 'median',
dashboard = TRUE, titvRaw = FALSE)
出图如下:
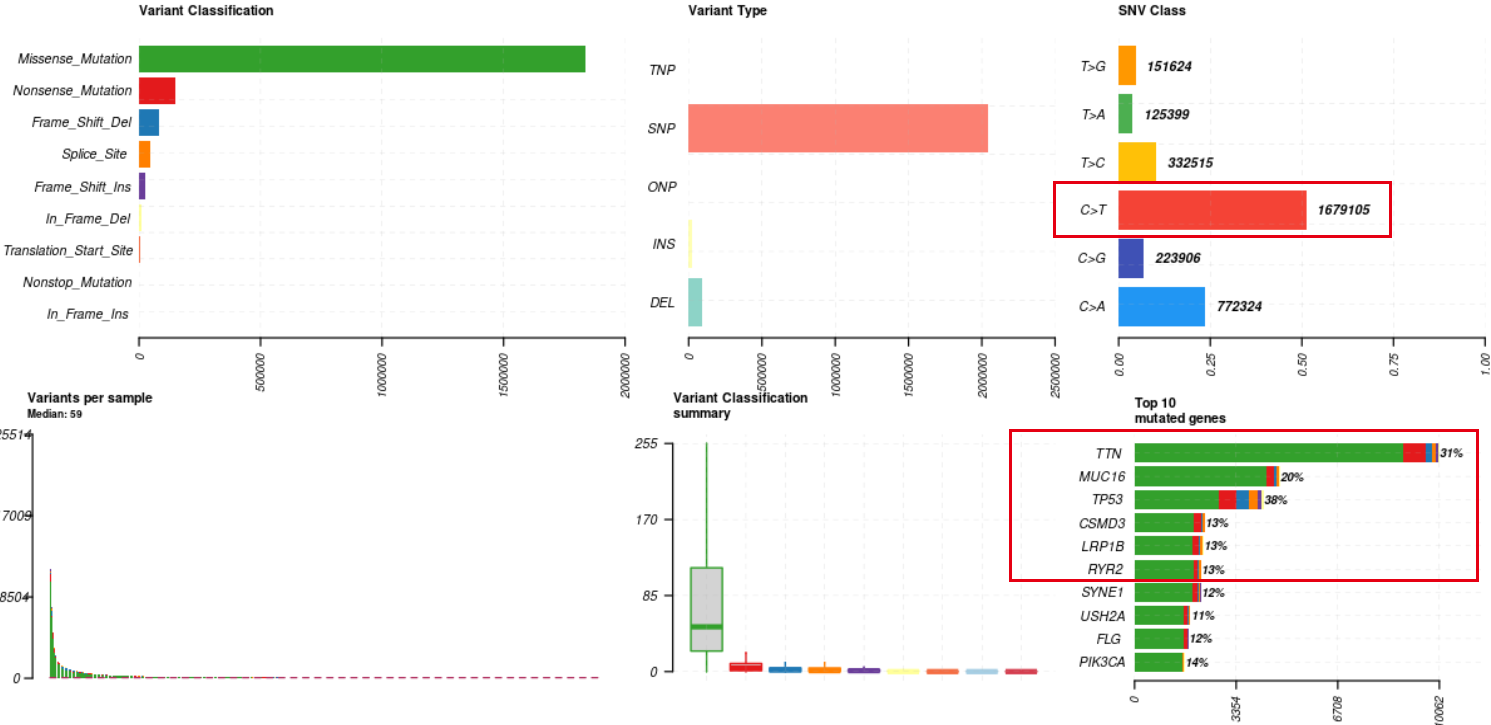
可以看到在TCGA队列的一万多个病人里面,突变比较多的基因是TTN,MUC16以及TP53,前面的两个基因好像是仅仅是因为基因比较长或者说基因上面的碱基特征问题导致肿瘤外显子的二代测序技术本身假阳性比较高。但是TP53确实是实打实的癌症相关基因了。同时大家也注意到了,其实比较出名的癌症基因大多并不是在top10里面,而且我们也不会关心全部的TCGA队列的一万多个病人,通常是挑选感兴趣的基因和病人去展示!
简单代码举例如下:
rm(list = ls())
require(maftools)
options(stringsAsFactors = F)
library(data.table)
tmp=fread('TCGA.BRCA.mutect.somatic.maf.gz')
cg=read.table('gene.txt')[,1] # 基因名字
cp=read.table('patient.txt')[,1] # 病人的ID
cg_tmp=tmp[tmp$Hugo_Symbol %in% cg,]
cg_cp_tmp=cg_tmp[substring(cg_tmp$Tumor_Sample_Barcode,1,15) %in% cp,]
laml = read.maf(maf = cg_cp_tmp)
oncoplot(maf = laml, top = 10) #高频突变的前10个基因,如下图所示
oncoplot(maf = laml,top = 10,draw_titv = TRUE) #在oncoplot中叠加titv图
上面的代码我没办法给大家3个文件,所以不能演示,不过我们可以针对tcga数据进行同样的操作。
首先过滤基因
这里建议根据自己的生物学背景进行挑选基因,比如铁死亡基因集,细胞焦亡基因集等等:
rm(list = ls())
require(maftools)
load(file = 'mc3.Rdata')
Genes = c(
"TP53",
"WT1",
"PHF6",
"DNMT3A",
"DNMT3B",
"TET1",
"TET2",
"IDH1",
"IDH2",
"FLT3",
"KIT",
"KRAS",
"NRAS",
"RUNX1",
"CEBPA",
"ASXL1",
"EZH2",
"KDM6A"
)
tmp=laml@data
cg_tmp=tmp[tmp$Hugo_Symbol %in% Genes,]
批量进行每个癌症内部绘图
真实代码,需要下载tcga数据库配对的临床信息 : Clinical with Follow-up - clinical_PANCAN_patient_with_followup.tsv ,链接是: http://api.gdc.cancer.gov/data/0fc78496-818b-4896-bd83-52db1f533c5c
或者 TCGA-Clinical Data Resource (CDR) Outcome - TCGA-CDR-SupplementalTableS1.xlsx :https://api.gdc.cancer.gov/data/1b5f413e-a8d1-4d10-92eb-7c4ae739ed81
library(readxl)
phe=read_excel('TCGA-CDR-SupplementalTableS1.xlsx',sheet = 1)[,2:3]
phe=as.data.frame(phe)
head(phe)
> head(phe)
bcr_patient_barcode type
1 TCGA-OR-A5J1 ACC
2 TCGA-OR-A5J2 ACC
3 TCGA-OR-A5J3 ACC
4 TCGA-OR-A5J4 ACC
5 TCGA-OR-A5J5 ACC
6 TCGA-OR-A5J6 ACC
接下来就是各个癌症批量出图:
table(phe$type)
cp_list=split(phe$bcr_patient_barcode,phe$type)
lapply(1:length(cp_list), function(i){
cp=cp_list[[i]]
pro=names(cp_list)[i]
cg_cp_tmp=cg_tmp[substring(cg_tmp$Tumor_Sample_Barcode,1,12) %in% cp,]
laml = read.maf(maf = cg_cp_tmp)
pdf(paste0('choose-genes-in-',pro,'.pdf'),height = 5)
oncoplot(maf = laml , removeNonMutated =F) #高频突变的前10个基因
dev.off()
})
简单看了看,这些基因在COAD里面突变比较多:
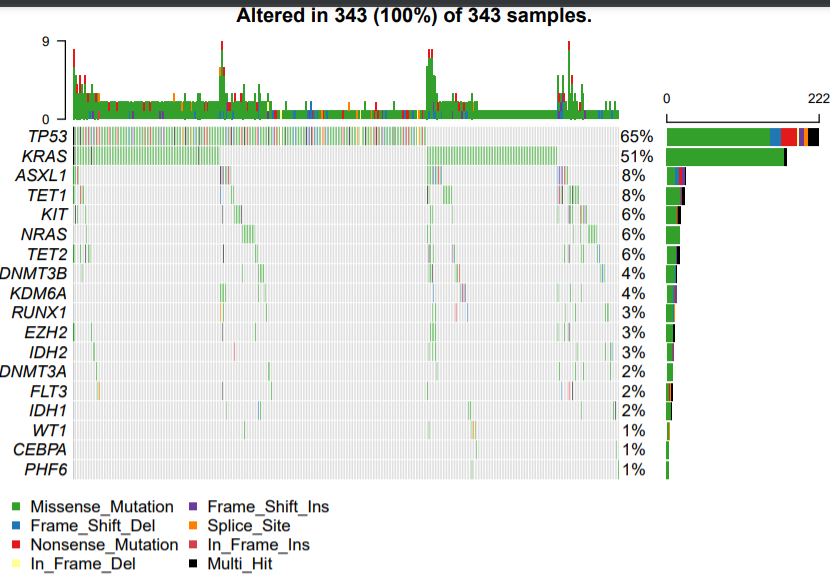
但是LUAD和LUASC,虽然都是肺癌,但是缺很不一样哦!
首先是LUAD:
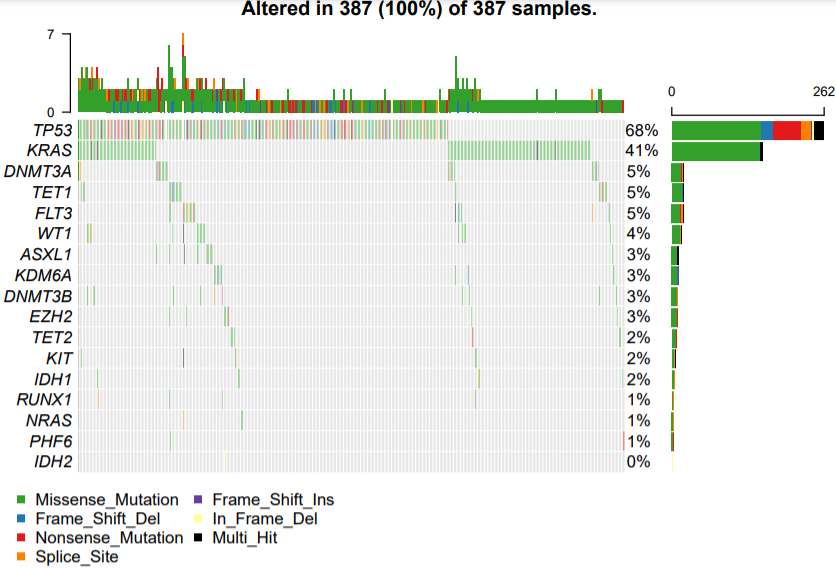
下面是LUSC:
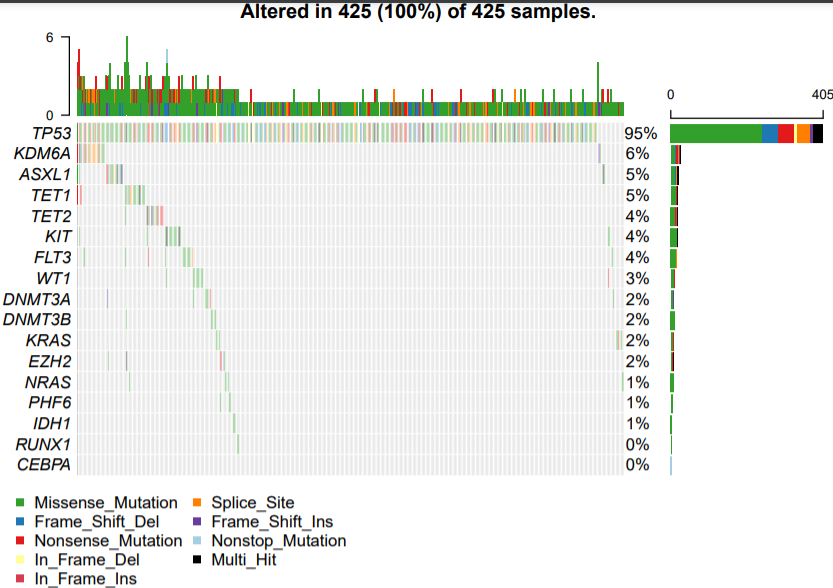
突变信息可以做的分析太多了,比如 :
才发现居然都是四五年前的教程了!恰好最近友军在开设相关课程:肿瘤基因组生物信息学培训班2021年唯一场次
开课时间:预计在10.10开课(如有其他情况,会至少提前一周通知)。
每周六、周日下午3:00~5:00上课:每课时45min,两节连上,中间休息15min。
课表如下:
这个课程是高阶课程,需要有一些linux及编程基础才可以。
如果您确实对这个课程感兴趣,但是没有编程及linux基础的话,我们可以提供我们自己录制的相关视频课程进行自学python及linux课程。对于无基础学员,通过我们的视频的学习与练习大约一周内就可以跟上课程要求了。
本课程结束后,您会有以下几方面的收获:
-
掌握肿瘤变异calling与注释的基本方法,对于影响变异的特殊因素及其解决方案有一定的认识。
-
掌握肿瘤免疫NGS相关分析,学会肿瘤免疫治疗的队列景观分析的研究方法。
-
掌握肿瘤克隆与进化的研究方法与手段,解析肿瘤队列挖掘关键基因,结合数据库进行解读以发表高水平研究的套路与方法。
-
适当拓展肿瘤NGS多组学研究的方法与手段。
-
最终可实现初步具备开展肿瘤变异研究的相关课题或者搭建与开发肿瘤相关应用产品的能力。
报名方式:
详见微信公众号的课程介绍:肿瘤基因组生物信息学培训班2021年唯一场次