关于这个estimate,我们在生信技能树公众号已经是多次分享了,主要是因为肿瘤本身具有异质性而且肿瘤取样问题,所以我们拿到了肿瘤数据(比如表达量矩阵)里面除了恶性癌症细胞的,还有基质细胞和免疫细胞的特性。所以2013年的一个数据挖掘文章,就整理了两个基因集来根据表达量矩阵去量化肿瘤样品里面的基质细胞和免疫细胞的比例。(其实是非常的粗糙,只不过是发的比较早,所以引用很多)
就是一个estimate包的estimate函数,使用起来是非常简单,大家可以自行查看包的函数的帮助文档:
# estimateScore {estimate} 函数
# StromalScorenumeric scalar specifying the presence of stromal cells in tumor tissue
# ImmuneScorenumeric scalar specifying the level of infiltrating immune cells in tumor tissue
# ESTIMATEScorenumeric scalar specifying tumor cellularity
# TumorPuritynumeric scalar specifying ESTIMATE-based tumor purity with value in range[0,1]
step1: 设置 estimate 的包和函数
首先安装一些必备的包:
rm(list=ls())
options(stringsAsFactors = F)
library(stringr)
library(data.table)
if(!require("estimate")){
library(utils)
rforge <- "http://r-forge.r-project.org"
install.packages("estimate", repos=rforge, dependencies=TRUE)
}
library(estimate)
library(stringr)
然后设置 estimate 的包和函数 ,这里针对RNA-seq数据,我们创造性的使用logCPM这样的表达量矩阵,进行estimate流程(必然与tpm或者fpkm矩阵结果不一致哦!)
# 参考:http://www.bio-info-trainee.com/6602.html
###### step1: 设置 estimate 的包和函数 ######
estimate_RNAseq <- function(RNAseq_logCPM,pro){
input.f=paste0(pro,'_estimate_input.txt')
output.f=paste0(pro,'_estimate_gene.gct')
output.ds=paste0(pro,'_estimate_score.gct')
write.table(RNAseq_logCPM,file = input.f,sep = '\t',quote = F)
library(estimate)
filterCommonGenes(input.f=input.f,
output.f=output.f ,
id="GeneSymbol")
estimateScore(input.ds = output.f,
output.ds=output.ds,
platform="illumina")
scores=read.table(output.ds,skip = 2,header = T)
rownames(scores)=scores[,1]
scores=t(scores[,3:ncol(scores)])
scores=data.frame(scores)
return(scores)
}
dir.create('estimate_results')
如果是根据具体某个基因表达量把病人分组,那么tpm或者fpkm跟我们的logCPM值并不会有区别,就是说差异分析,生存分析都没有问题。
但是呢,现在我们做的是estimate分析,它本质上是ssGSEA,这个分析依赖于同一个样品内部基因的排序,所以tpm或者fpkm的归一化会改变样品内基因的排序,所以结果会有差异!
step2: 一个个癌症内部运行estimate
首先 前面的教程 不同癌症的差异难道大于其与正常对照差异吗,以及 大样本量多分组表达量矩阵分析你难道没想到单细胞吗,我们认识了TCGA数据库的33种癌症的全部的表达量矩阵,并且格式化保存为了 Rdata 文件。
接下来就是载入这些 Rdata 文件文件,各个癌症内部独立运行 estimate分析函数:
###### step2: 一个个癌症内部运行estimate ######
fs=list.files('Rdata/',pattern = 'htseq_counts')
fs
# 首先,针对全部的TCGA数据库癌症的表达量矩阵批量运行 estimateScore {estimate} 函数
# StromalScorenumeric scalar specifying the presence of stromal cells in tumor tissue
# ImmuneScorenumeric scalar specifying the level of infiltrating immune cells in tumor tissue
# ESTIMATEScorenumeric scalar specifying tumor cellularity
# TumorPuritynumeric scalar specifying ESTIMATE-based tumor purity with value in range[0,1]
lapply(fs, function(x){
# x=fs[1]
pro=gsub('.htseq_counts..Rdata','',x)
print(pro)
load(file = file.path('Rdata/',x))
dat=log2(edgeR::cpm(pd_mat)+1)
scores=estimate_RNAseq(dat,pro)
head(scores)
scores$group = ifelse(
as.numeric(substring(rownames(scores),14,15)) < 10,
'tumor','normal'
)
table(scores$group )
})
这个包,非常的弱智,居然需要输出两个gct文件和一个txt文件,不过这里我们选择尊重作者,不修改它函数。
批量计算完毕后,就可以 批量载入各自 estimate 结果 进行绘图,代码如下:
fs=list.files('estimate_results/',
pattern = 'estimate_RNAseq-score-forTCGA')
fs
lapply(fs, function(x){
# x=fs[1]
pro=gsub('.Rdata','',
gsub('estimate_RNAseq-score-forTCGA-','',x))
print(pro)
load(file = file.path('estimate_results/',x))
#可视化
library(ggplot2)
library(ggpubr)
library(patchwork)
library(ggsci)
library(ggstatsplot)
# 省略
})
每个癌症内部所有的样品都被estimate算法计算了3个值:
-
StromalScorenumeric scalar specifying the presence of stromal cells in tumor tissue
-
ImmuneScorenumeric scalar specifying the level of infiltrating immune cells in tumor tissue
- ESTIMATEScorenumeric scalar specifying tumor cellularity
各种各样的可视化
可视化绘图代码实在是太长了,这里略,总之我们拿到了全部的样品的estimate结果,包括肿瘤样品和配对正常组织样品的,33个癌症都有!
也可以直接 针对前面的seurat对象运行 estimate !
###### step4: 针对前面的seurat对象运行 estimate ######
library(Seurat)
load(file = 'sce.Rdata')
gp=substring(colnames(sce),14,15)
table(gp)
sce@meta.data$gp=gp
pd_mat=sce@assays$RNA@counts
dat=log2(edgeR::cpm(pd_mat)+1)
pro='estimate_for_seurat'
scores=estimate_RNAseq(dat,pro)
head(scores)
scores$group= sce@meta.data$gp
scores$type= sce@meta.data$group
save(scores,file = file.path('estimate_results',
paste0('estimate_RNAseq-score-for-',
pro,'.Rdata')))
所有的可视化都是针对于 scores 这个数据框 ,存储在 estimate_RNAseq-score-for-estimate_for_seurat.Rdata 这个文件里面 (我也不知道直接为什么把它文件名搞这么复杂了):
> head(scores)
StromalScore ImmuneScore ESTIMATEScore group type
TCGA.OR.A5JP.01A -997.6522 -1415.33255 -2412.9848 01 ACC
TCGA.OR.A5JG.01A -1115.8320 -962.83539 -2078.6674 01 ACC
TCGA.OR.A5K1.01A -904.5518 -671.54985 -1576.1017 01 ACC
TCGA.OR.A5JR.01A -1191.8237 -670.32161 -1862.1453 01 ACC
TCGA.OR.A5KU.01A -1171.0190 -1483.48861 -2654.5076 01 ACC
TCGA.OR.A5L9.01A -460.7720 79.30757 -381.4645 01 ACC
> tail(scores)
StromalScore ImmuneScore ESTIMATEScore group type
TCGA.V4.A9EU.01A -1296.493 -865.5307 -2162.024 01 UVM
TCGA.WC.A87T.01A -1839.759 -1736.4595 -3576.219 01 UVM
TCGA.WC.AA9A.01A -1954.846 -1738.1963 -3693.042 01 UVM
TCGA.V4.A9EA.01A -1430.072 -1190.5262 -2620.598 01 UVM
TCGA.RZ.AB0B.01A -1293.130 -1194.0290 -2487.159 01 UVM
TCGA.V4.A9F8.01A -1048.983 -860.7518 -1909.735 01 UVM
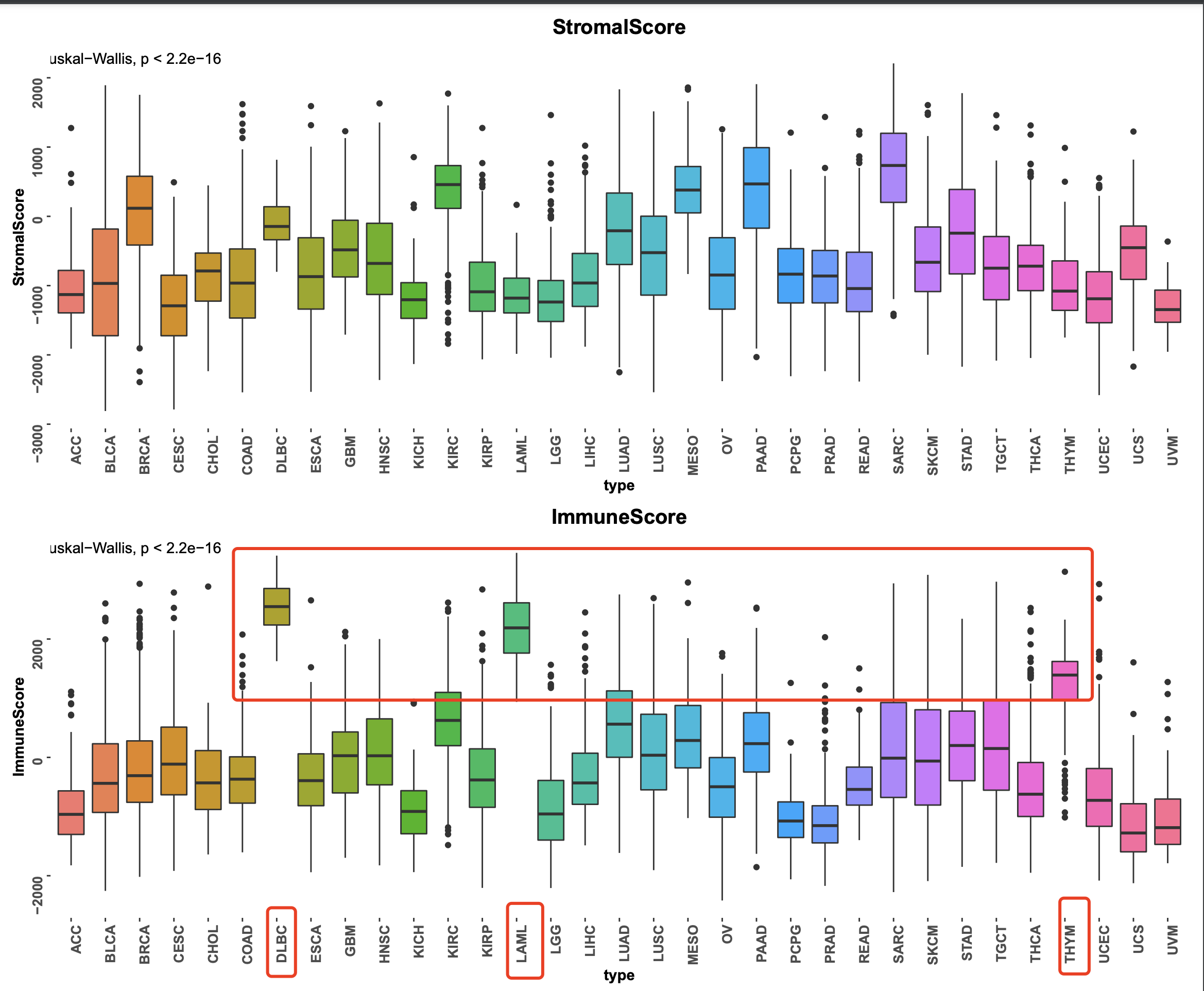
不同分组的estimate的两个打分值比较
可以看到,03,代表Primary Blood Derived Cancer - Peripheral Blood, 它不是实体肿瘤,是血液瘤, 对应LAML 这个癌症,它的免疫打分是最高的,合情合理。

如果具体去看肿瘤样品里面的estimate的两个打分值比较,就会发现,除了LAML之外,还有DLBC和THYM两个癌症的免疫打分超级高!
当然了,基质打分高的癌症也值得关注,每个人生物学背景不一样,可以自行看图!
那么,问题来了,肿瘤病人的正常对照组织的estimate的两个打分值有没有生物学意义呢?

任何数据分析结果都需要辅助合理的生物学解释,但是这个estimate算法本身就是一个很粗糙的,虽然可以根据表达量矩阵去量化肿瘤样品里面的基质细胞和免疫细胞的比例,但是可信度也是存疑。