对指定基因进行干扰,然后看它造成的全局转录水平的影响已经成为了探索该基因功能的主流思路。这也就是为什么转录组测序技术成为了大众首选,一个最简单的项目只需要2个分组合起来6个样品即可,算起来不到5000块钱人民币,如果分析的好就是一个独立的课题!
而对基因的干扰,其实有正向和反向两个路线,就是敲除一个基因以及过表达它。以我们朴素的思维来说,这两个完全相反的干扰设计理论上会造成起码是相反的效果!但实际情况下,在不同场景下干扰一个基因其实也会效果迥然不同。
先看看下面的两个文章,前者是在非小细胞肺癌里面过表达KIAA0101这个基因,后者是在白血病里面敲除KIAA0101基因 :
- 发表在 J Cancer 2020;的文章:《Overexpression of KIAA0101 Promotes the Progression of Non-small Cell Lung Cancer》,链接是:https://www.jcancer.org/v11p6663.htm
- 发表在 Ann Transl Med. 2021 Mar; 的文章:《KIAA0101 knockdown inhibits cell proliferation and induces cell cycle arrest and cell apoptosis in chronic lymphocytic leukemia cells》,链接是:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8039647/
在非小细胞肺癌里面过表达KIAA0101
这个文章 超级简单,就是去肺腺癌和肺鳞癌的公共数据集去说明KIAA0101这个基因确实是癌症部位的表达量显著的高于癌旁,而且它基本上可以完美的区分癌症和癌旁,虽然它高低表达量划分来区分病人的生存不一定会很好。
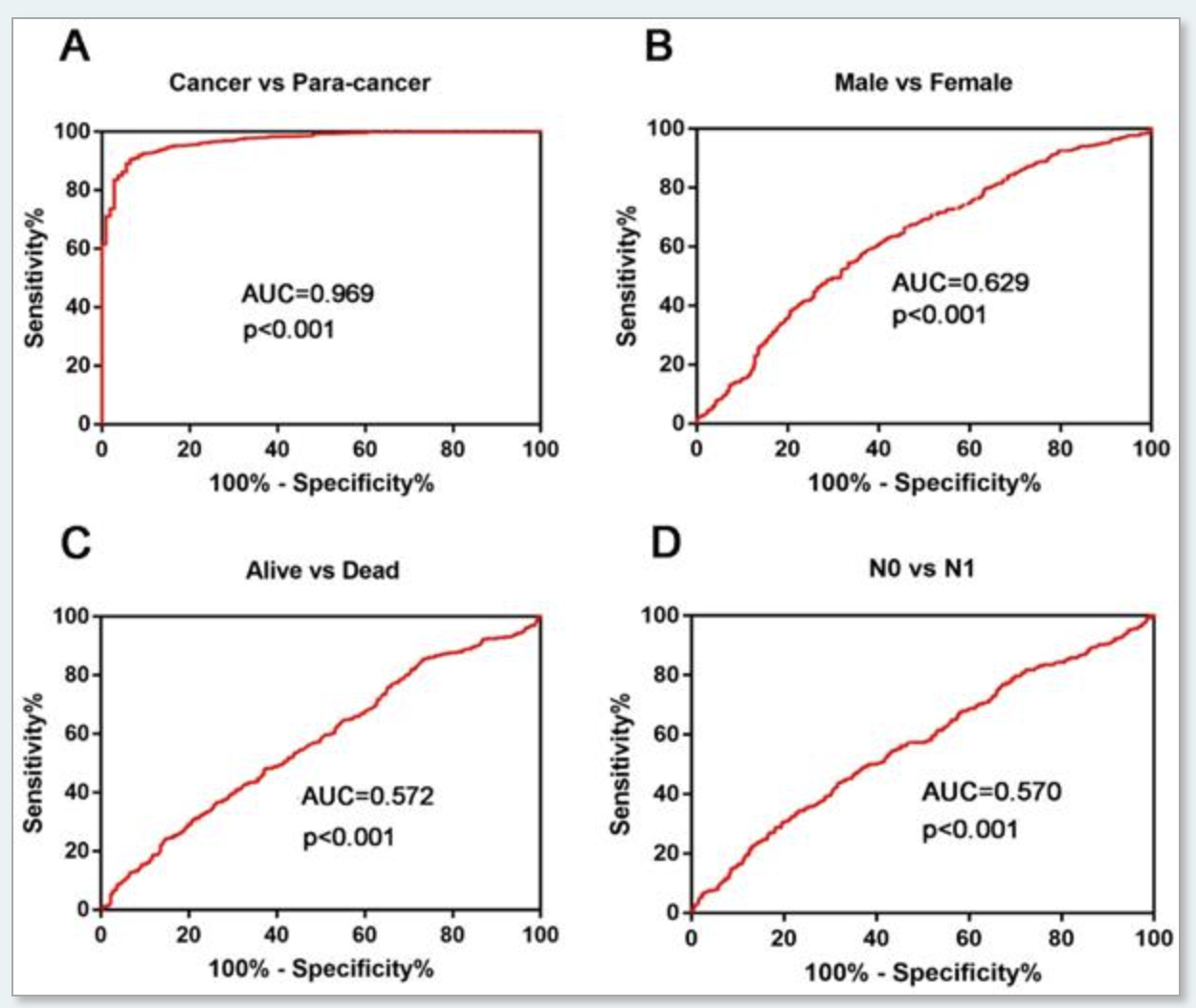
可以看到拿 KIAA0101这个基因 表达量区分病人生存情况,跟区分病人性别一样的的没有效果啊, 甚至都没有办法区分淋巴结转移与否这么重要的临床事件。
需要理解肺癌的生物学背景知识,组织病理上通常将肺癌分为 - 非小细胞肺癌(non-small-cell lung cancer,NSCLC)
- 小细胞肺癌(small cell lung cancer,SCLC)
其中SCLC约占全部肺癌的15%~20%,SCLC的发病与吸烟密切相关,生物学特征为分化程度低、恶性程度高、倍增时间快、侵袭性强、预后差,中位生存期才7个月左右。
其中NSCLC又可以区分为LUSC和LUAD,已经是鳞癌和腺癌的差异。
虽然这个研究里面作者并没有给出 它自己的数据,但是正文里面列出来了,Identification of KIAA0101-related pathways with GSEA. High KIAA0101-related genes significantly enriched in (A) TP53 signaling pathway and (B) cell cycle.
也就是说,作者的数据方面结果是 在非小细胞肺癌里面过表达KIAA0101这个基因,发现TP53和cell cycle这两个信号通路是上调的, 也就是说激活了癌症相关通路。然后它选择了几个肺癌细胞系,The knockdown of KIAA0101 expression inhibited NSCLC migration in vitro.
仍然是非常朴素的思维,认为这个KIAA0101基因啊,就是一个坏的东西,它促进了癌症发生发展,所以治疗癌症就是去抑制它即可。白血病里面敲除KIAA0101基因
同样的,作者发现了这个KIAA0101基因在疾病里面上调 :
- the mRNA expression pattern of KIAA0101 in CD19+ B cells isolated from the peripheral blood of 6 healthy donors and 26 CLL patients was examined through RT-qPCR.
所以作者选择干扰降低KIAA0101基因 表达:Affymetrix GeneChip analysis was performed to identify differentially expressed genes in Granta-519 cells infected with shKIAA0101 lentiviruses versus cells infected with shctrl lentiviruses.
通过简单的差异分析,设置阈值: (|fold change| >1.5, P<0.05). 得到了:(253 up-regulated, 532 down-regulated) ,然后作者基本上就是在瞎搞,把全部的基因拿去生物学功能数据库富集一下:
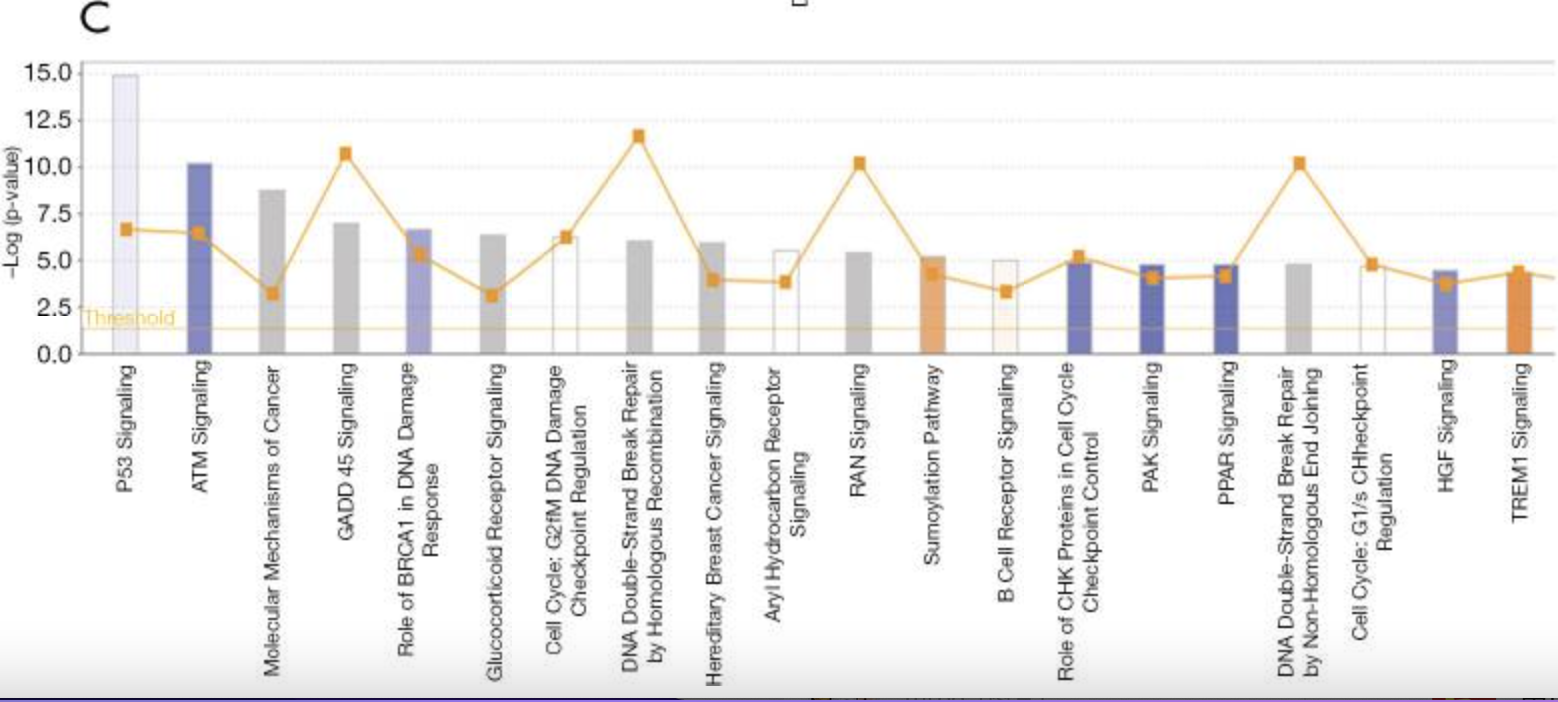
有意思的是这个数据集也是不提供的!那我们该如何去对比说明过表达一个基因和敲除它的作用一定是相反的吗?
让我们做一个数学假设,你有同一个病人的癌症样品9份,其中3份你过表达了某基因,另外3份你敲除了该基因,这样的9份样品送去公司做完转录组后,你对这个表达量矩阵做2次差异分析发现:
- 过表达组相当于对照组,上下调各自基因列表
- 敲除组相当于对照组,上下调各自基因列表
这个时候你有4个基因列表,如果你做交集 - 某些基因在两次差异分析都是上调
- 某些基因在两次差异分析都是下调
- 某些基因在过表达组是上调,在敲除组是下调
- 某些基因在过表达组是下调,在敲除组是上调
你会如何解释这4个基因集?
如果你也需要转录组测序,欢迎关注我们的转录组产品线: - 明码标价之转录组常规测序服务(仅需799每个样品)
- 明码标价之普通转录组上游分析
- 明码标价之转录组下游分析仅需800元
- 明码标价之转录组测序数据的可变剪切
- 明码标价之RNA-Seq数据的内含子保留分析
以及 公共数据库挖掘 产品线 - 明码标价之公共数据库的生存分析
- 明码标价之公共数据集的WGCNA
- 明码标价之公共数据库探索
- 明码标价之探索新流程(以MSIpred为例)
- 明码标价之任意科研图表绘制(以氨基酸的位点变异图为例)