差异分析,大家都喜欢两个分组的比较,但实际科研项目,往往是比这复杂,多达十几个甚至几十个分组也不稀奇。之前的教程:[多分组的差异分析只需要合理设置design矩阵即可](https://mp.weixin.qq.com/s/cnBLsAFWOimrxVnfnYEIDw),我们展示了无论多少个分组,都可以很方便的进行差异分析。
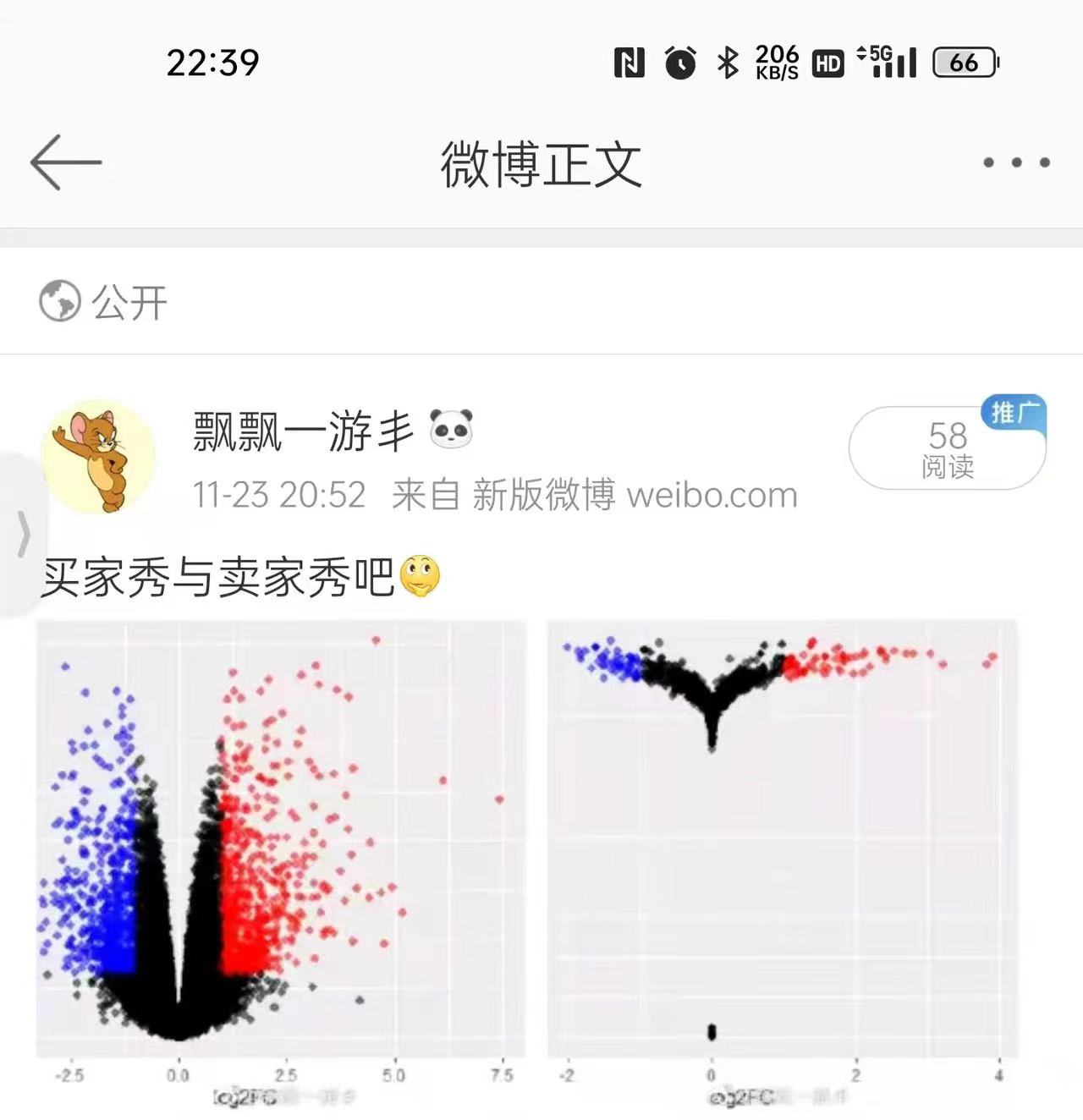
对两个分组的差异分析,我们会首选火山图进行展示,但是有一个学员反馈了他画出来的一个超级诡异的火山图,我把它命名为烟花图!
下面我们就以 airway 这个R包来演示这个过程,如果你没有这个包,就自己使用代码 :BiocManager::install(‘airway’) 进行安装哦!
整理数据
# 1.构建表达矩阵 ----------------------------------------------------------------
dir.create("./data")
# 魔幻操作,一键清空
rm(list = ls())
options(stringsAsFactors = F)
# BiocManager::install('airway')
# 加载airway数据集并转换为表达矩阵
library(airway,quietly = T)
data(airway)
class(airway)
rawcount <- assay(airway)
colnames(rawcount)
# 查看表达谱
rawcount[1:4,1:4]
# 去除前的基因表达矩阵情况
dim(rawcount)
# 获取分组信息
group_list <- colData(airway)$dex
group_list
# 过滤在至少在75%的样本中都有表达的基因
keep <- rowSums(rawcount>0) >= floor(0.75*ncol(rawcount))
table(keep)
filter_count <- rawcount[keep,]
filter_count[1:4,1:4]
dim(filter_count)
# 加载edgeR包计算counts per millio(cpm) 表达矩阵,并对结果取log2值
library(edgeR)
express_cpm <- log2(cpm(filter_count)+1)
express_cpm[1:6,1:6]
# 保存表达矩阵和分组结果
save(filter_count,
express_cpm,
group_list,
file = "./data/Step01-airwayData.Rdata")
代码块里面的内容有点多, 实际上仅仅是一个熟悉airway这个表达量矩阵的过程:
正常差异分析
我们这里首先选取最出名的包,DESeq2来进行差异分析:
#2.进行差异分析 ------------------------------------------------------------------
rm(list = ls())
options(stringsAsFactors = F)
# 读取基因表达矩阵信息
lname <- load(file = "./data/Step01-airwayData.Rdata")
lname
# 查看分组信息和表达矩阵数据
exprSet <- filter_count
dim(exprSet)
exprSet[1:6,1:6]
table(group_list)
# 加载包
library(DESeq2)
# 第一步,构建DESeq2的DESeq对象
colData <- data.frame(row.names=colnames(exprSet),group_list=group_list)
dds <- DESeqDataSetFromMatrix(countData = exprSet,colData = colData,design = ~ group_list)
# 第二步,进行差异表达分析
dds2 <- DESeq(dds)
# 提取差异分析结果,trt组对untrt组的差异分析结果
tmp <- results(dds2,contrast=c("group_list","trt","untrt"))
DEG_DESeq2 <- as.data.frame(tmp[order(tmp$padj),])
head(DEG_DESeq2)
# 去除差异分析结果中包含NA值的行
DEG_DESeq2 = na.omit(DEG_DESeq2)
# 筛选上下调,设定阈值
fc_cutoff <- 1
fdr <- 0.05
DEG_DESeq2$regulated <- "normal"
loc_up <- intersect(which(DEG_DESeq2$log2FoldChange>log2(fc_cutoff)),
which(DEG_DESeq2$padj<fdr))
loc_down <- intersect(which(DEG_DESeq2$log2FoldChange< (-log2(fc_cutoff))),
which(DEG_DESeq2$padj<fdr))
DEG_DESeq2$regulated[loc_up] <- "up"
DEG_DESeq2$regulated[loc_down] <- "down"
table(DEG_DESeq2$regulated)
head(DEG_DESeq2)
library(AnnoProbe)
ag=annoGene(rownames(DEG_DESeq2),
ID_type = 'ENSEMBL',species = 'human'
)
head(ag)
DEG_DESeq2$ENSEMBL=rownames(DEG_DESeq2)
deg_anno=merge(ag,DEG_DESeq2,by='ENSEMBL')
head(deg_anno)
# 保存
write.table(deg_anno,"./data/DEG_DESeq2_all.xls",
row.names = F,sep="\t",quote = F)
save(deg_anno, file = "./data/Step03-DESeq2_nrDEG.Rdata")
得到了上下调基因,有了差异分析结果表格,每个表格都可以去判断统计学显著的上下调基因,去富集分析,去做火山图,热图。这样的分析看我六年前的表达芯片的公共数据库挖掘系列推文即可哈 :
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
不过我们这里仅仅是展现火山图即可:
绘制火山图
## 首先检查是否上下调设置错了
# 挑选一个差异表达基因
load(file = "./data/Step03-DESeq2_nrDEG.Rdata")
load(file = "./data/Step01-airwayData.Rdata")
head(deg_anno)
# 在差异基因列表里面,随意挑选一个上下调基因
# 这里上调基因举例说明
cg="ENSG00000152583"
deg_anno[match(cg, deg_anno$ENSEMBL),]
exp <- c(t(express_cpm[match(cg,rownames(express_cpm)),]))
test <- data.frame(value=exp,group=group_list)
library(ggplot2)
library(ggpubr)
df <- data.frame(value=exp,group=group_list)
head(df)
p <- ggboxplot(df, x = "group", y = "value",
color = "group", palette = "jco",
add = "jitter")
p
# Add p-value
p + stat_compare_means()
library(EnhancedVolcano)
colnames(deg_anno)
EnhancedVolcano(deg_anno,
lab = deg_anno$SYMBOL,
x = 'log2FoldChange',
y = 'padj')
ggsave(filename = 'EnhancedVolcano_for_DEG_DEseq2.pdf')
如下所示:
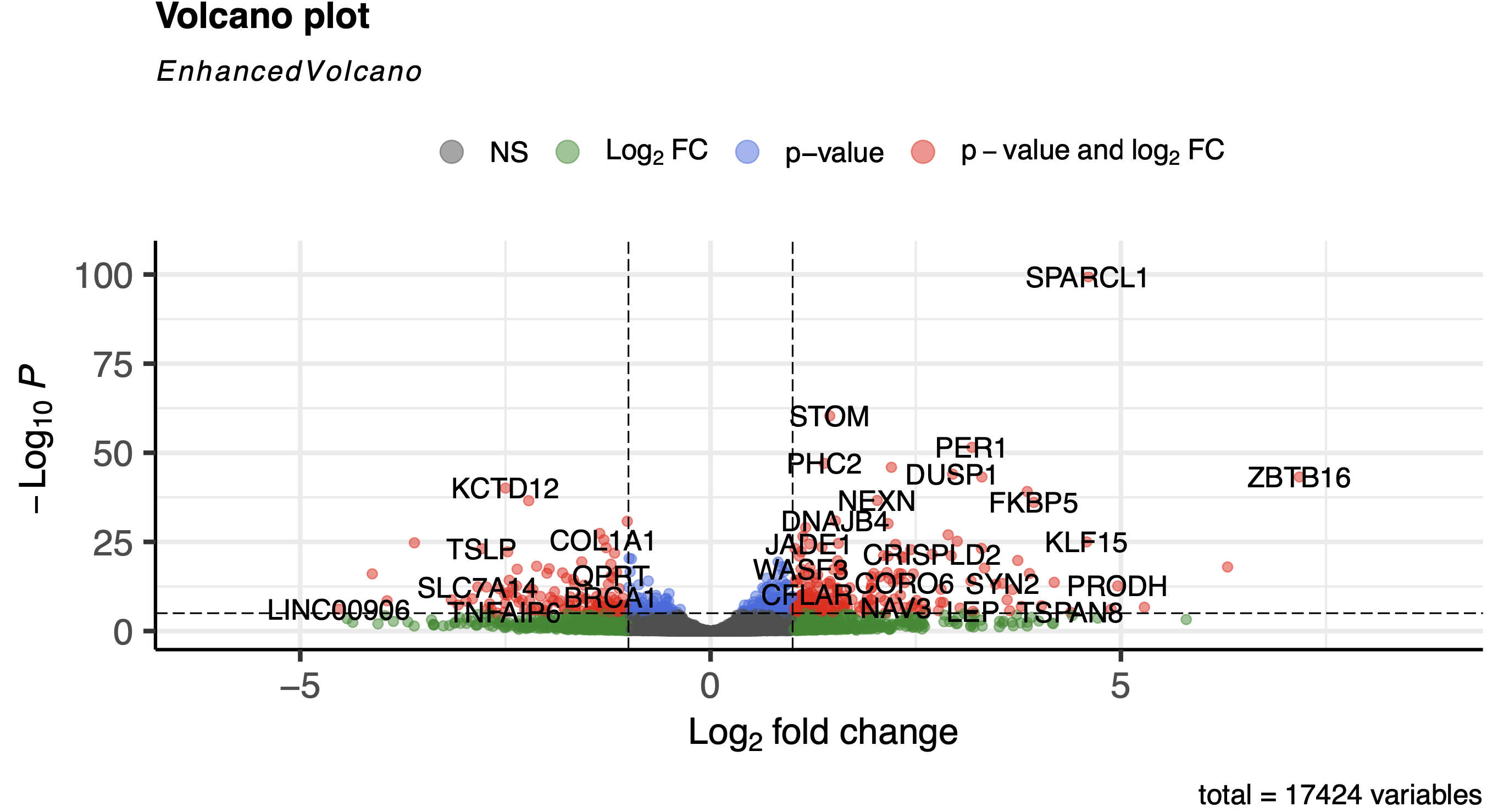
创作烟花图所需要的数据
火山图是基于正常的表达量矩阵进行正常的差异分析得到的:
> exprSet[1:4,1:4]
SRR1039508 SRR1039509 SRR1039512 SRR1039513
ENSG00000000003 679 448 873 408
ENSG00000000419 467 515 621 365
ENSG00000000457 260 211 263 164
ENSG00000000460 60 55 40 35
> group_list
[1] untrt trt untrt trt untrt trt untrt trt
Levels: trt untrt
> table(group_list)
group_list
trt untrt
4 4
但是对于烟花图,我们需要把前面的两个分组的两个样品进行重复四次,假装是每个分组有4个重复值,实际上每个分组里面的重复都是人造的。
代码如下所示:
rm(list = ls())
options(stringsAsFactors = F)
# 读取基因表达矩阵信息
lname <- load(file = "./data/Step01-airwayData.Rdata")
lname
# 查看分组信息和表达矩阵数据
exprSet <- filter_count
dim(exprSet)
exprSet[1:4,1:4]
group_list
table(group_list)
# 加载包
library(DESeq2)
exprSet=cbind(
exprSet[,1:2],
exprSet[,1:2],
exprSet[,1:2],
exprSet[,1:2]
)
colnames(exprSet)=1:8
# DESeq2包不受欺骗,不支持我们的这样的假冒伪劣矩阵哦
if(F){
# 加载包
library(DESeq2)
# 第一步,构建DESeq2的DESeq对象
colData <- data.frame(row.names=colnames(exprSet),group_list=group_list)
dds <- DESeqDataSetFromMatrix(countData = exprSet,colData = colData,design = ~ group_list)
# 第二步,进行差异表达分析
dds2 <- DESeq(dds)
# 提取差异分析结果,trt组对untrt组的差异分析结果
tmp <- results(dds2,contrast=c("group_list","trt","untrt"))
DEG_DESeq2 <- as.data.frame(tmp[order(tmp$padj),])
head(DEG_DESeq2)
}
# 加载包
library(limma)
library(edgeR)
#创建设计矩阵和对比:假设数据符合分布,构建线性模型
design <- model.matrix(~0+factor(group_list))
colnames(design) <- levels(factor(group_list))
rownames(design) <- colnames(exprSet)
design
#设置需要进行对比的分组
comp <- 'trt-untrt'
cont.matrix <- makeContrasts(contrasts=c(comp),levels = design)
#进行差异表达分析
dge <- DGEList(counts=exprSet)
v <- voom(dge,design,plot=TRUE, normalize="quantile")
fit <- lmFit(v, design)
fit2 <- contrasts.fit(fit,cont.matrix)
fit2 <- eBayes(fit2)
tmp <- topTable(fit2, coef=comp, n=Inf,adjust.method="BH")
DEG_limma_voom <- na.omit(tmp)
head(DEG_limma_voom)
library(AnnoProbe)
ag=annoGene(rownames(DEG_limma_voom),
ID_type = 'ENSEMBL',species = 'human'
)
head(ag)
DEG_limma_voom$ENSEMBL=rownames(DEG_limma_voom)
deg_anno=merge(ag,DEG_limma_voom,by='ENSEMBL')
head(deg_anno)
library(EnhancedVolcano)
colnames(deg_anno)
EnhancedVolcano(deg_anno,
lab =deg_anno$SYMBOL,
x = 'logFC',
y = 'adj.P.Val')
如下所示:
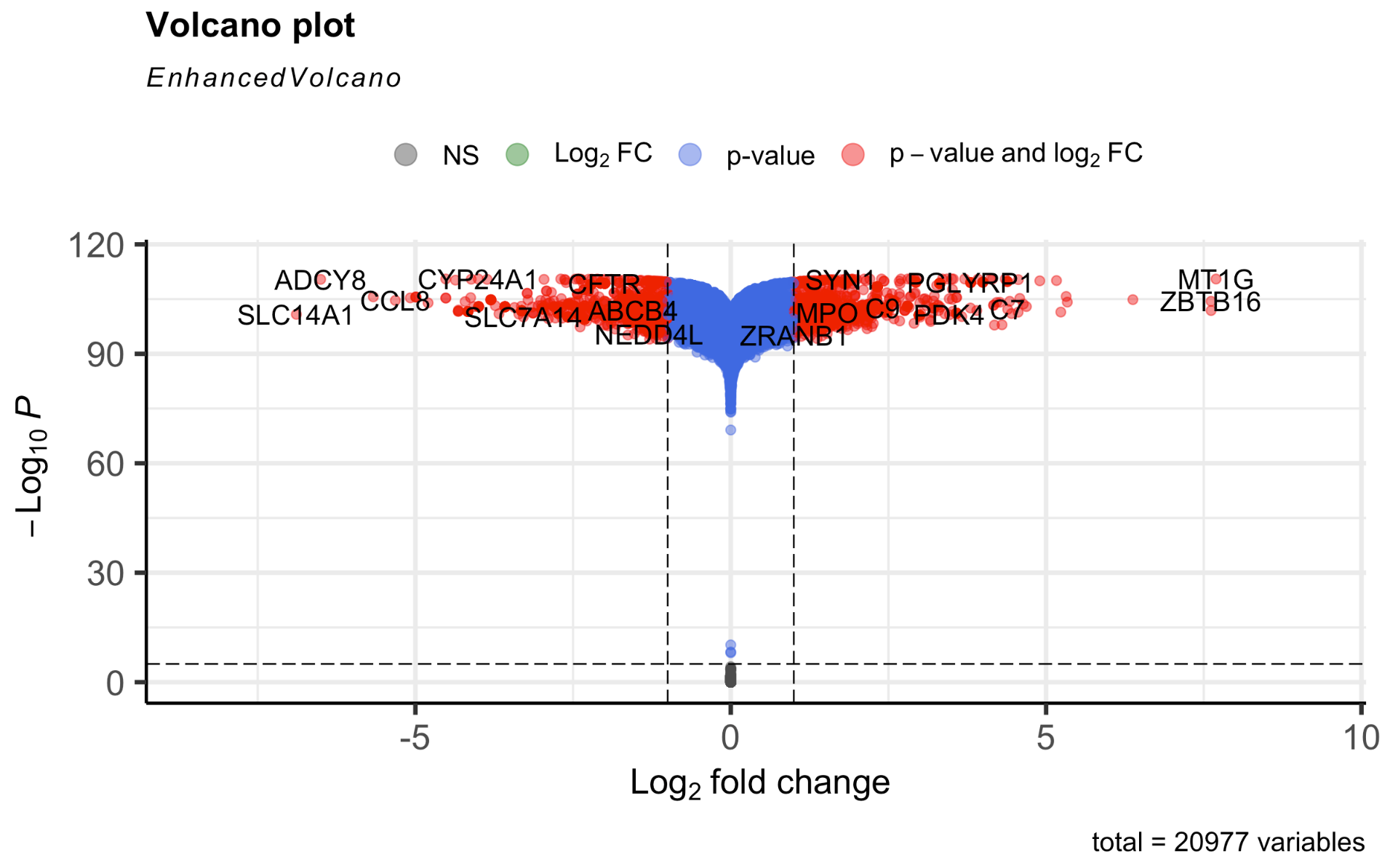
学徒作业:
运行我这个教程里面的代码,得到两次差异分析结果后,进行比较,绘制各自上下调基因的韦恩图看看!
思考题
既然我们前面把每个分组里面的单个样品简单粗暴的复制粘贴3次充当生物学重复,会导致计算得到的p值如此的诡异。
那么, 假如你的两个分组真的就是都有且仅有一个样品,你该如何进行差异分析呢?