前面我们已经成功完成了《CNS图表复现专辑》的前20个关键图表的复现,现在开启第二波图表复现,会大量触及到补充图表。 目录及前面的代码总结 CNS图表复现专辑第二波开启 。
如果是现在才看到这个系列的小伙伴建议自己去先读一下CELL杂志的文章:Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing ,因为作者提供了全套代码,在:https://github.com/czbiohub/scell_lung_adenocarcinoma ,研究者们共收集到30位患者的49份活检样本(biopsy),分为三种类型:治疗前(TKI naive [TN]),靶向治疗后肿瘤消退或稳定(RD, residual disease state)以及靶向治疗后肿瘤仍然增长(PD, upon subsequent progressive disease),这样单细胞转录组数据就非常丰富!
在 CNS图表复现专辑第二波开启 可以看到前面的降维聚类分群,就是不知道为什么T和B这样的淋巴细胞和髓系都是有轻微混入了,而且它们跟上皮细胞居然在umap上面并不是泾渭分明的。难道是我因为我前面没有针对样品或者病人进行整合吗?我前面对 在 CNS图表复现专辑第二波开启 教程里面的指出来smart-seq2 技术的单细胞, CCA整合并没有必要 ,主要是因为每个样品其实就几百个细胞而已,并不是10X技术那样的每个样品都是好几千个细胞。
我们把一个肿瘤单细胞转录组数据进行初步降维聚类分群,并且各个单细胞亚群独立保存成为了seurat对象,接下来就很容易去抽取T和B淋巴细胞对象里面的表达量矩阵作为从单细胞转录组数据推断肿瘤拷贝数的正常二倍体参考细胞,然后对上皮细胞表达量矩阵进行肿瘤拷贝数推断!
rm(list=ls())
library(Seurat)
library(ggplot2)
library(clustree)
library(cowplot)
library(dplyr)
sce.all=readRDS( "2-int/sce.all_int.rds")
sce.all
table(sce.all@meta.data$seurat_clusters)
load(file = '3-cell/phe_by_markers.Rdata')
table(phe$celltype)
sce.all$celltype=phe$celltype
sce.all.list <- SplitObject(sce.all , split.by = "celltype")
sce.all.list
for (i in names(sce.all.list)) {
epi_mat = sce.all.list[[i]]@assays$RNA@counts
epi_phe = sce.all.list[[i]]@meta.data
sce=CreateSeuratObject(counts = epi_mat,
meta.data = epi_phe )
sce
table(sce@meta.data$orig.ident)
save(sce,file = paste0(i,'.Rdata'))
}
得到的文件如下所示;
21M 8 19 16:46 B.Rdata
9.2M 8 19 16:46 Mast.Rdata
61M 8 19 16:46 T.Rdata
18M 8 19 16:46 endo.Rdata
161M 8 19 16:46 epi.Rdata
58M 8 19 16:46 fibo.Rdata
101M 8 19 16:45 myeloid.Rdata
另外,请不要再找我要这些Rdata文件了,但凡是你看完了前面的 CNS图表复现专辑第二波开启 教程,这些代码跑一下就自己制作出来了全部的数据文件!
其实我们在教程:CNS图表复现09—上皮细胞可以区分为恶性与否 提到了五千多个上皮细胞里面只有三千七百左右是恶性细胞,但是 copykat 和 infercnv这两个从单细胞转录组数据推断肿瘤拷贝数变异技术差异还没有被探索过。
构建两个算法都需要的输入数据
其中 infercnv 算法需要3个文件,但是 copykat 只需一个文件即可,我们这里一起制作。
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
load(file = 'first_sce.Rdata') # 这里面仅仅是上皮细胞哦
epiMat=as.data.frame(GetAssayData( sce , slot='counts',assay='RNA'))
load('../Rdata-celltype/B.Rdata') # 这里面仅仅是B淋巴 细胞哦
B_cellMat=as.data.frame(GetAssayData( sce, slot='counts',assay='RNA'))
load('../Rdata-celltype/T.Rdata') # 这里面仅仅是T淋巴 细胞哦
T_cellsMat=as.data.frame(GetAssayData( sce, slot='counts',assay='RNA'))
B_cellMat=B_cellMat[,sample(1:ncol(B_cellMat),800)]
T_cellsMat=T_cellsMat[,sample(1:ncol(T_cellsMat),800)]
dat=cbind(epiMat,B_cellMat,T_cellsMat)
groupinfo=data.frame(v1=colnames(dat),
v2=c(rep('epi',ncol(epiMat)),
rep('spike-B_cell',300),
rep('ref-B_cell',500),
rep('spike-T_cells',300),
rep('ref-T_cells',500)))
library(AnnoProbe)
geneInfor=annoGene(rownames(dat),"SYMBOL",'human')
colnames(geneInfor)
geneInfor=geneInfor[with(geneInfor, order(chr, start)),c(1,4:6)]
geneInfor=geneInfor[!duplicated(geneInfor[,1]),]
length(unique(geneInfor[,1]))
head(geneInfor)
## 这里可以去除性染色体
# 也可以把染色体排序方式改变
dat=dat[rownames(dat) %in% geneInfor[,1],]
dat=dat[match( geneInfor[,1], rownames(dat) ),]
dim(dat)
expFile='expFile.txt'
write.table(dat,file = expFile,sep = '\t',quote = F)
groupFiles='groupFiles.txt'
head(groupinfo)
write.table(groupinfo,file = groupFiles,sep = '\t',quote = F,col.names = F,row.names = F)
head(geneInfor)
geneFile='geneFile.txt'
write.table(geneInfor,file = geneFile,sep = '\t',quote = F,col.names = F,row.names = F)
首先走inferCNV流程
有了前面的3个文件,接下来走inferCNV流程超级简单!真正的核心代码就 是 infercnv::run 函数,来源于 infercnv 这个包!
代码如下所示:
options(stringsAsFactors = F)
library(Seurat)
library(gplots)
library(ggplot2)
expFile='expFile.txt'
groupFiles='groupFiles.txt'
geneFile='geneFile.txt'
library(infercnv)
infercnv_obj = CreateInfercnvObject(raw_counts_matrix=expFile,
annotations_file=groupFiles,
delim="\t",
gene_order_file= geneFile,
ref_group_names=c("ref-B_cell",'ref-T_cells')) ## 这个取决于自己的分组信息里面的
# cutoff=1 works well for Smart-seq2, and cutoff=0.1 works well for 10x Genomics
infercnv_obj2 = infercnv::run(infercnv_obj,
cutoff=0.1, # cutoff=1 works well for Smart-seq2, and cutoff=0.1 works well for 10x Genomics
out_dir= "infercnv_output", # dir is auto-created for storing outputs
cluster_by_groups=F, # cluster
hclust_method="ward.D2", plot_steps=F)
得出如下所示的结果:
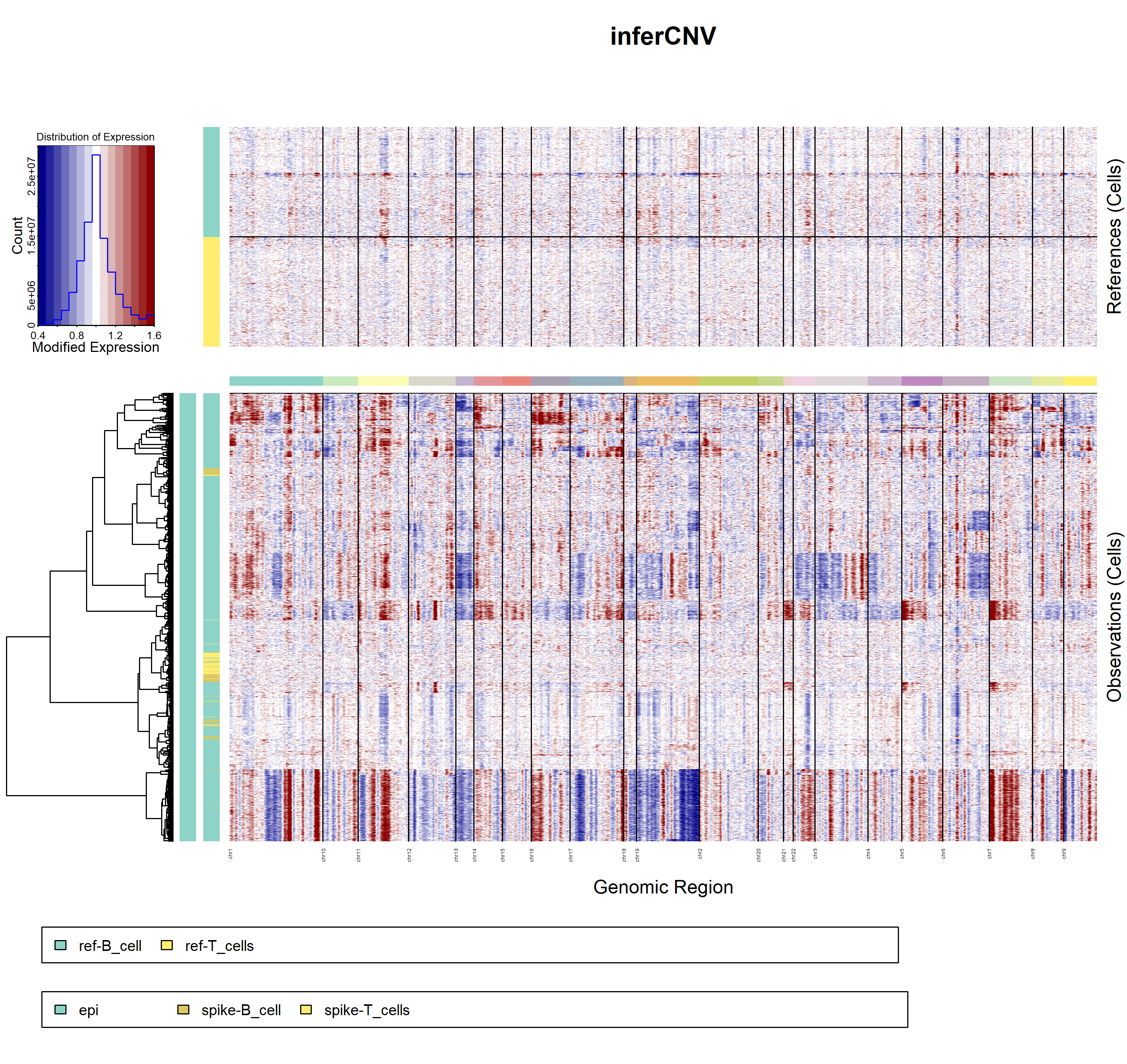
可以很容易看出来,我们特意加进去的normal细胞确实是被判定为了normal细胞,与此同时,约三分之一的上皮细胞也会被判定为normal细胞 。跟前面的教程:CNS图表复现09—上皮细胞可以区分为恶性与否 提到了五千多个上皮细胞里面只有三千七百左右是恶性细胞,是一致的。
然后走copykat流程
有了前面的3个文件,接下来走copykat流程超级简单!真正的核心代码就 是 copykat 函数,来源于 copykat这个包!
代码如下:
options(stringsAsFactors = F)
library(Seurat)
library(gplots)
library(ggplot2)
expFile='expFile.txt'
groupFiles='groupFiles.txt'
geneFile='geneFile.txt'
library(copykat)
exp.File <- read.delim( expFile , row.names=1)
exp.File[1:4,1:4]
groupFiles <- read.delim( groupFiles , header=FALSE)
head(groupFiles)
head( colnames(exp.File))
colnames(exp.File)=gsub('^X','', colnames(exp.File))
colnames(exp.File)=gsub('[.]','-', colnames(exp.File))
head( colnames(exp.File))
table(groupFiles$V2)
normalCells <- colnames(exp.File)[grepl("ref",groupFiles$V2)]
head(normalCells)
Sys.time()
res <- copykat(rawmat=exp.File, # 准备好的表达量矩阵
id.type="S", # 选择 symbol,因为我们的表达量矩阵 里面是它
ngene.chr=5,
win.size=25,
KS.cut=0.1,
sam.name="test", # 输出的一系列文件的前缀
distance="euclidean",
norm.cell.names=normalCells, # 正常细胞的名字
n.cores=1,
output.seg="FLASE")
Sys.time()
save(res,file = 'ref-of-copykat.Rdata')
可以看到:
> Sys.time()
[1] "2021-12-02 10:14:49 CST"
# 本身 copykat 函数运行过程也会自己记录时间
[1] "running copykat v1.0.5 updated 07/15/2021"
[1] "step1: read and filter data ..."
[1] "22493 genes, 6830 cells in raw data"
[1] "12355 genes past LOW.DR filtering"
[1] "step 2: annotations gene coordinates ..."
[1] "start annotation ..."
[1] "step 3: smoothing data with dlm ..."
[1] "step 4: measuring baselines ..."
[1] "896 known normal cells found in dataset"
[1] "run with known normal..."
[1] "baseline is from known input"
[1] "step 5: segmentation..."
[1] "step 6: convert to genomic bins..."
[1] "step 7: adjust baseline ..."
[1] "step 8: final prediction ..."
[1] "step 9: saving results..."
[1] "step 10: ploting heatmap ..."
Time difference of 4.091698 hours
> Sys.time()
[1] "2021-12-02 14:20:19 CST"
可以看到,不到7000个细胞还是需要运行4个小时之久,不过也是可以通过设置 线程数量 来加快这个进度!很不幸的是这个功能在Windows系统平台不支持:
Error in parallel::mclapply(1:ncol(norm.mat), dlm.sm, mc.cores = n.cores) :
'mc.cores' > 1 is not supported on Windows
不过,如果你也可以通过拆分样品的然后并行全部的样品的方式进行曲线救国,也算是并行计算:
ls -d S*|while read id;do
echo $id;
cd $id;
nohup Rscript.exe ../scripts/step4-run-copykat.R &
cd ../
done
比如你在每个 样品文件夹(以S开头的文件夹)里面都存放 文件名是 expFile.txt 的矩阵
任何就可以制作一个脚本: step4-run-copykat.R 进行如上所示的并行啦。
这样的话每个病人运行十几分钟即可,超级快!
然后我们一起看看copykat针对全部的病人混合群里的结果吧!

很神奇的是,这个算法对上皮细胞的恶性判断有点宽松啊,我们肉眼看到的大量的非整倍体的恶性细胞,居然都被算法给漏掉了。被这个算法定义为恶性上皮细胞的居然只有不到一千个细胞 :
aneuploid diploid
epi 851 3965
ref-B_cell 1 411
ref-T_cells 0 480
spike-B_cell 1 228
spike-T_cells 0 288
确实好奇怪啊,因为非常多的其它细胞我们肉眼看起来也是很恶性的,应该是 aneuploid,不知道为什么会被这个算法错误的判断为 diploid ,可能是我们的这个数据集是smart-seq2,并不是常见的10x数据集?